【www.zhangdahai.com--其他范文】
刘晓颖 蔡 斌 甘 露 金诗炜 蒋瑾瑾 周 霖
假肥大型肌营养不良包括杜氏肌营养不良(Duchenne muscular dystrophy,DMD)和贝氏肌营养不良(Becker muscular dystrophy,BMD),均是由抗肌萎缩蛋白(dystrophin)的基因突变所致的X连锁隐性遗传病。DMD常为致死性,临床表现为进行性加重的肌萎缩及肌无力,活产男婴的发病率为1/6 000~1/3 500。患者常在35岁出现明显症状,部分患者20岁左右可因心、肺功能衰竭而死亡[1-2]。BMD患者的临床过程与DMD相似,但病情进展缓慢,预后良好。通过对假肥大型肌营养不良先证者及其家系成员进行DMD基因突变分析,可以协助临床症状不典型患儿的早期诊断。现回顾性分析海军军医大学第一附属医院儿科近年收治的5例假肥大型肌营养不良患儿资料,以加强对该疾病的认识。
1.1 研究对象 回顾性分析2009年7月-2020年5月收治于海军军医大学第一附属医院儿科,并确诊为假肥大型肌营养不良的5例患儿资料。患儿监护人均签署知情同意书。
1.2 诊断标准 假肥大型肌营养不良诊断标准参考2016年《中国假肥大型肌营养不良症诊治指南》[1]。患儿临床症状不典型时,若出现不明原因的心肌酶谱指标水平升高、DMD基因突变,亦可支持诊断。
1.3 研究方法 收集5例患儿的一般资料、临床症状、体征(主要为Gower征)、实验室检查结果、肌肉活组织检查(简称活检)结果等。实验室检查的主要指标包括肌酸激酶(creatine kinase, CK)、肌酸激酶同工酶(creatine kinase isoenzyme MB, CK-MB)、ALT、AST、乳酸脱氢酶(lactate dehydrogenase, LDH)等,其正常值范围分别为CK 25~200 U/L,CK-MB<25 U/L,ALT<64 U/L,AST<64 U/L, LDH 80~285 U/L。
1.4DMD基因检测方法 采集患儿及其父母外周静脉血2 mL,应用高通量测序进行DMD全基因panel检测。全基因检查长度2.2 Mb,检测内容包括缺失重复及点突变(外显子+内含子)。DMD基因缺失(或重复)的验证采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)。

2.1 一般资料分析 纳入的5例假肥大型肌营养不良患儿均为男性,就诊时年龄范围为2 d~8岁,发病年龄范围为1 d~4岁。主要首发表现为双下肢无力、步态不稳、皮肤黄染、肝酶水平升高、新生儿感染等。病例1、3和4均因“肝酶异常”曾至外院就诊,并因误诊只行保肝治疗。病例2的姨表兄曾罹患肌营养不良(具体不详),余患儿均无家族史。病例4和5因年幼无法配合完成Gower征的体格检查,余3例患儿Gower征均为阳性(+),并伴有双侧腓肠肌肥大。病例1完成肌肉活检,符合肌营养不良改变,肌电图结果提示肌源性损害导致的肌电改变。见表1。

表1 5例患儿一般资料
2.2 实验室检查结果 5例患儿CK水平均显著升高,伴CK-MB、ALT、AST和LDH水平不同程度的升高。5例患儿CK、CK-MB、ALT、AST、LDH水平分别为(15 810±10 378) U/L、(351.2±212.4) U/L、(202.6±149.9) U/L、(202.4±82.0) U/L、(1 068.0±583.8) U/L,分别为正常上限的 22.9~162.8倍、5.7~28.0倍、0.8~6.9倍、1.6~5.3倍、1.8~6.6倍。分别比较CK升高倍数与CK-MB、ALT、AST和LDH升高倍数,差异均有统计学意义(P=0.000 5或0.001 6),后4者升高倍数之间两两比较的差异均无统计学意义(P值均>0.05)。见表2。

表2 5例患儿实验室检查结果 (U/L)
2.3 基因检测结果 因病例1、2原始基因报告难以获得,本研究详述病例3、4、5基因检测结果,分别见图1、图2、图3。


A 病例3患儿的基因检测结果提示患儿DMD基因49~52号外显子半合子缺失,为自发突变 B 病例3患儿父亲的基因检测结果提示该区域无变异 C 病例3患儿母亲的基因检测结果提示该区域无变异图1 病例3 DMD基因检测结果(图上方序列号为样本编号)

A 病例4患儿的基因检测结果提示患儿DMD基因45号外显子半合子缺失,为自发突变 B 病例4患儿父亲的基因检测结果提示该区域无缺失 C 病例4患儿母亲的基因检测结果提示该区域无缺失图2 病例4 DMD基因检测结果(图上方序列号为样本编号)
4例患儿基因检测提示DMD基因缺失突变,其中自发突变3例,遗传自母亲突变1例,突变位点分别位于48~51号、42~43号、49~52号、45号外显子,既往均见报道,诊断明确。病例5患儿基因检测结果为DMD基因单纯57~59外显子重复突变,为自发突变,既往数据库中未见此突变报道。见表3。

表3 5例患儿基因检测结果
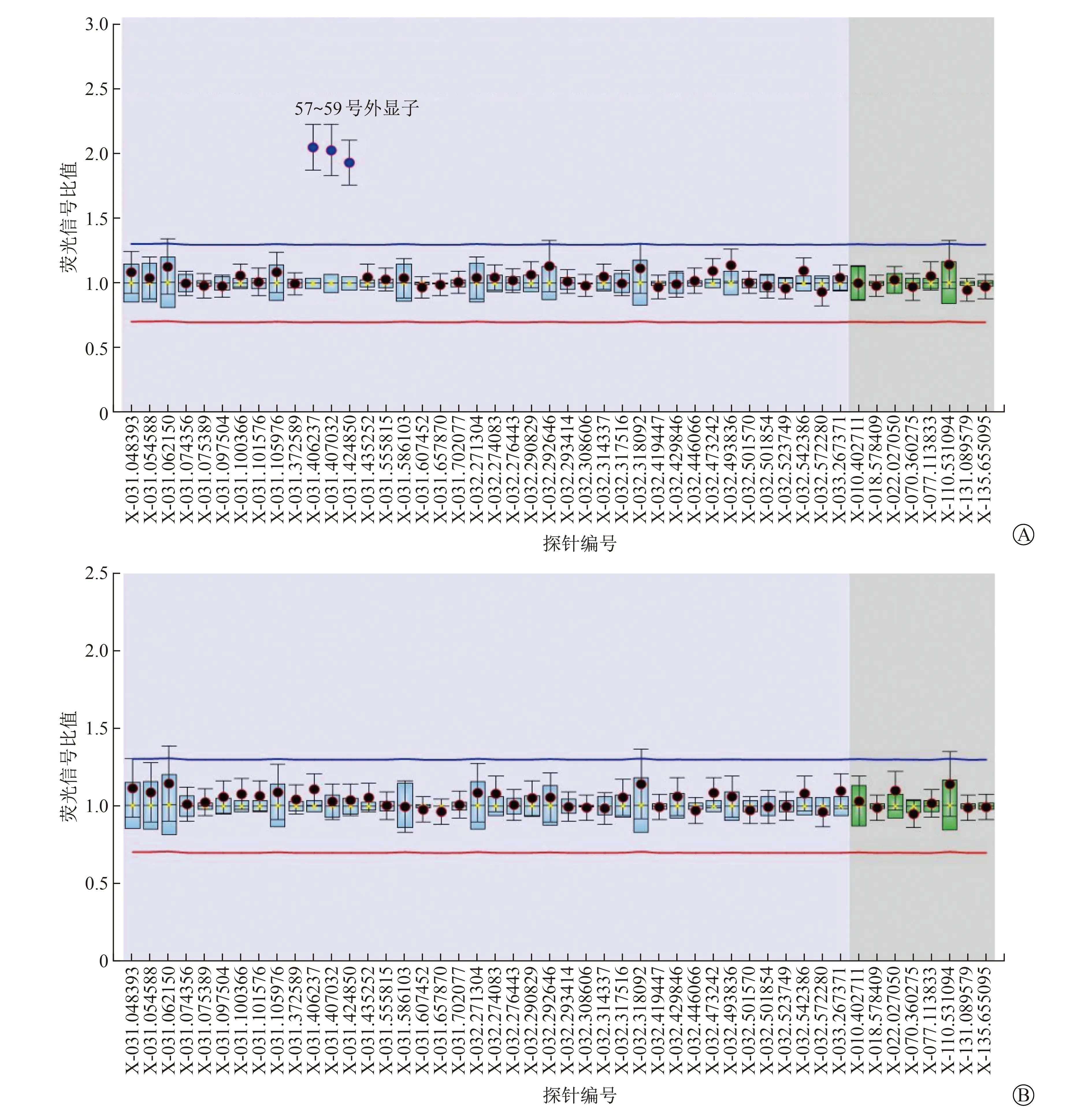
A 病例5患儿MLPA分析结果提示患儿DMD基因57~59号外显子重复变异,为自发突变 B 病例5患儿母亲MLPA分析结果提示无此变异 (横坐标代表探针编号及检测基因-所在外显子;
纵坐标代表患者与对照样本荧光信号的比值,红蓝线为基准线,比值高于蓝线为重复,比值低于红线为缺失,荧光信号强度介于0.7~1.3为正常)图3 病例5患儿DMD基因MLPA分析
假肥大型肌营养不良是由位于Xp21.1的DMD基因缺陷所致的X连锁隐性遗传病。DMD基因突变若影响了其开放阅读框,蛋白质表达则被过早截断,引发DMD;
突变若保留了开放阅读框,蛋白质的功能则被部分保留,引发BMD。故与BMD相比,DMD发病早、预后差,更受学者及临床医师的关注。本文仅针对DMD进行讨论。
不同年龄的DMD患儿具有不同的临床特征,诊断要点包括:①X连锁隐性遗传,3~5岁隐袭起病,进行性发展,一般在12岁后出现行走障碍;
②可出现双下肢无力、鸭步、Gower征、起蹲困难和腓肠肌肥大等临床表现;
③血清CK水平显著升高至正常值的数十倍至上百倍;
④肌电图显示肌源性受损;
⑤肌肉活检呈典型的肌源性损伤表现,且抗肌萎缩蛋白抗体免疫组织化学染色呈阴性;
⑥超声心动图可显示左心室扩大,肌肉MRI提示肌肉水肿和脂肪浸润;
⑦DMD基因检测为外显子缺失、重复、微小突变或点突变[1]。在以上7条诊断标准中,年幼儿肌电图和肌肉活检的接受度较差,不易实施;
心脏超声和肌肉MRI则不能直接提供诊断线索。故在临床实践中,临床症状和CK检测结果是较易获得的重要的诊断依据。典型特殊姿势和步态包括Gower征,常于患儿幼年发病,但起病隐匿,可无典型临床表现,直至患儿20岁左右因呼吸、心力衰竭就诊,但DMD发展迅速,一经诊断疾病常已达中晚期。因此,在疾病早期,患儿的症状常常被忽视,易误诊、漏诊。
本研究5例患儿均于5岁前起病,病例3、4为婴儿期起病,病例5为新生儿期起病,目前关于婴儿及新生儿起病的报道较少。因年幼患儿在行Gower征检查时不能配合,为诊断带来一定难度,故部分患儿常在发病数年后才得以确诊[3]。在实验室检查方面,患儿除了有典型的CK水平升高,亦伴有心肌酶及肝酶谱指标的异常,临床上易漏诊或误诊为“心肌炎”“肝功能异常”等[4-5]。虽然,AST和ALT水平升高通常提示肝细胞损伤,但肌肉是血清转氨酶的潜在来源。肌营养不良、炎性肌病和代谢性肌病等肌肉疾病可导致血液中肌酸磷酸激酶(CPK)、LDH、ALT和AST水平升高,当伴有肝功能异常、肝酶水平升高时,需考虑到儿童肌肉疾病。当遇到有肝酶水平升高而没有肝病相关临床症状或体征的患儿时,应尽早检测CK水平,有助于肌肉疾病的早期诊断,并避免类似肝活检等不必要的侵入性检查[6-10]。年龄≤3岁的患儿CK水平升高10倍以上时应考虑DMD可能。本研究中5例患儿CK、CK-MB、ALT、AST、LDH水平均升高,但以CK升高尤为显著,提示CK可能是DMD最敏感的实验室检测指标,可用于区别肝脏与心肌损害。新生儿及婴儿期CK水平已显著升高,对于发病早、临床症状隐匿的DMD幼年患儿有重要的诊断意义。
此外,也有国内学者报道1例因肠道感染就诊发现心肌酶水平升高的年长患儿,其无肌肉萎缩、四肢无力、运动障碍、腓肠肌肥大,Gower征呈阴性,拟诊心肌炎并行营养心肌治疗,后经肌电图及基因检测确诊为DMD[6]。DMD患儿在左心室收缩功能下降之前可出现心肌损害,而心肌病是导致DMD患儿死亡的主要原因。本研究中5例患儿的CK-MB水平均明显升高(5.7~28.0倍),提示疾病早期即可出现CK-MB水平升高,应尽早完善相关检查,对DMD早期随访和干预有一定意义。
抗肌萎缩蛋白是目前所知DMD的唯一致病靶点。DMD基因突变多为大缺失、大重复和点突变[7]。本研究5例患儿中4例为自发基因突变,其中1例系遗传自母亲。基因突变类型80%为缺失突变,突变位点与既往文献报道一致, 基因突变分布于外显子两个热点区域(外显子44~53号和外显子2~20号缺失热区),与文献报道基本相符。病例5患儿基因检测结果为DMD基因单纯57~59号外显子重复突变,既往鲜见相关报道。曾有研究[8]显示,DMD基因52~62号外显子重复突变,与本研究中重复变异区域有包含关系。自发突变患儿母亲MLPA分析未发现DMD基因外显子拷贝数异常,但不能排除其母亲生殖腺镶嵌体突变的情况存在,亦无法确定变异发生阶段,因此仍建议患儿及其亲属行遗传咨询,以降低再发风险。本研究中5例患儿发病均较早,考虑假肥大型肌营养不良中DMD可能性较大,但DMD基因检测未体现是否破坏“开放阅读框”,因患儿年幼、症状隐匿、体格检查不能配合,仅根据目前的临床表现和实验室检查尚不能完全确诊为DMD,需进一步随访其临床表现以明确其类型。对疑诊假肥大型肌营养不良的患儿进行基因检测,通过其先证者及家系成员进行突变分析,可以发现未经文献及数据库报道的新突变,从而丰富DMD的基因突变谱。
探索DMD理想的生物标志物有助于DMD的诊断。肌特异性微核糖核酸(miRNA)在DMD患者的骨骼肌中富集,且在DMD发病机制中起重要作用,故有学者认为肌特异性miRNA可能成为理想的DMD生物标志物[9]。DMD分子诊断可用于临床诊断。对于依据临床表现和血生物化学检测结果疑诊DMD或BMD的患者,首先选择多重PCR扩增技术(multiplex polymerase chain reaction,mPCR)分析,阴性者行MLPA技术对DMD基因进行缺失及重复突变筛查,对MLPA检测阴性者采用二代测序(NGS)进行检测,如仍未发现异常,进行肌活检蛋白质分析和cDNA分析,这可能是最佳、最经济的基因诊断策略[5,10]。
因目前尚无DMD有效的根治方案,糖皮质激素是唯一被循证医学证实有效且可以延缓病程的药物。近年来,研究者探索治疗DMD的方法包括研发抑制炎症反应、调节钙水平、促进肌肉生长、抗纤维化、促进线粒体功能等针对肌营养不良机制的对症治疗药物,物理治疗(适量的运动训练、应用矫形支具、虚拟现实游戏),以及基因替代、外显子跳跃、基因编辑等基因治疗方案和干细胞治疗[11-13]。采用基因手段治疗和改善DMD症状、缩短病程、提高生活质量,将为DMD的有效治疗带来希望[14]。准确的患者和携带者基因检测和产前筛查结果,是降低DMD发病率的关键。
综上所述,对于不明原因肝酶、CK-MB水平升高的年幼患儿,应考虑DMD诊断。年幼患儿如不能配合神经系统体格检查,抗拒肌肉活检等有创检查,应尽早完善CK水平检测,并积极进行DMD基因检测,以实现早期诊断、早期治疗,减少漏诊及误诊。
猜你喜欢 外显子基因突变病例 外显子跳跃模式中组蛋白修饰的组合模式分析电子科技大学学报(2022年5期)2022-10-29外显子组测序助力产前诊断胎儿骨骼发育不良中国生殖健康(2020年4期)2021-01-18管家基因突变导致面部特异性出生缺陷的原因中国生殖健康(2020年2期)2021-01-18“病例”和“病历”作文评点报·低幼版(2020年25期)2020-07-23外显子组测序助力产前诊断胎儿骨骼发育不良中国生殖健康(2018年4期)2018-11-06基因突变的“新物种”小学生导刊(2018年13期)2018-06-29也门霍乱疫情更新中国感染与化疗杂志(2018年3期)2018-01-20管家基因突变导致面部特异性出生缺陷的原因中国生殖健康(2018年2期)2018-01-12先天性巨细胞病毒感染致connexin26基因突变新生儿听力随访及干预医学研究杂志(2015年12期)2015-06-10人类组成型和可变外显子的密码子偏性及聚类分析湖北农业科学(2014年11期)2014-09-10本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0716/626211.html