【www.zhangdahai.com--其他范文】
关 凡 吴晓敏 黄 河 曾朝阳 熊 炜 范春梅*
(1)中南大学基础医学院,长沙 410013;
2)中南大学肿瘤研究所,国家卫健委癌变原理重点实验室和教育部癌变与侵袭原理重点实验室,长沙 410078)
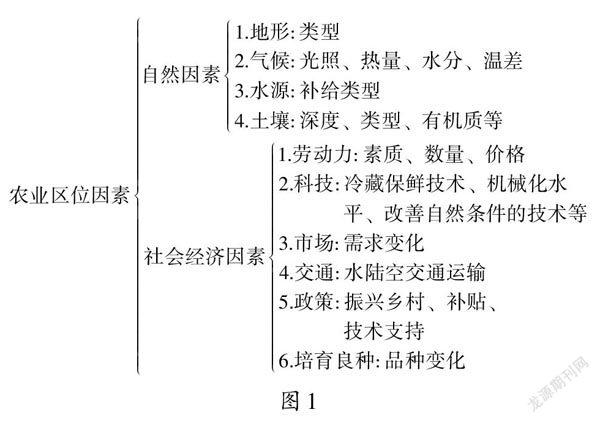
在过去的几十年里,随着DNA 测序技术的发展,越来越多的证据已经表明“走出非洲学说”比“多地区起源学说”更加经得起推敲。1987年,在科学家们的追踪下,发现20 万年前生活在非洲的一位妇女是全世界人的祖先。在这里,科学家们追踪的是婴儿的线粒体DNA(mtDNA),众所周知线粒体DNA 只能通过母亲传给下一代,所以科学家们最先发现的便是生活在远古时代的一位妇女,据此,便推测人类起源于非洲。为了排除亚洲独立起源假说,复旦大学校长金力及其团队[1]对来自中国各地的9 988 例男性随机样本进行了3个Y 染色体单倍型的基因分型,结果显示这些样本全部都携带有来自非洲的“基因痕迹”。这一证据直接否定了中国独立起源的假说。除此之外,金力也表示并不是所有走出非洲的原始人类都和现代人的起源有关系。美国华盛顿大学的生物学家Alan R.Templeton 通过研究单倍型树对人类基因流模式和历史事件进行了重建,发现在190万年前人类祖先发生了第一次从非洲出走,大约70 万年前发生了第二次,第三次出走发生在大约10 万年前[2](图1)。第二次出走的是尼安德特人,第三次出走的一批才是现代人的祖先,这两批人并不是毫无交集。随着技术的发展,科学家们通过基因组研究发现,尼安德特人与现代人的祖先在地球上相遇了,并且有了基因的交流。
2022年10 月3 日,瑞典卡罗琳斯卡医学院宣布,将2022年诺贝尔生理学或医学奖授予瑞典生物学家斯万特·帕博,以表彰他在已灭绝人类基因组和人类进化方面所做出的重要贡献。斯万特·帕博开创了一门新的学科——古人类基因组学,该学科的发展壮大不断完善“走出非洲学说”,同时在研究过程中也开发了许多新的技术,为更好地研究人类进化提供了技术支持。他从人口迁徙和种群杂交两个方面解释了现代人基因多样性的来源,从自然选择的角度诠释了尼安德特人遗留给现代人基因的生理意义,同时也为研究疾病指引了新的方向。

Fig.1 The ancients left Africa three times图1 古人类三次走出非洲
1.1 尼安德特人与丹尼索瓦人
作为第二批大规模走出非洲的人——尼安德特人,一直到1856年才正式被命名。1829年尼安德特人的头骨化石首次在比利时被发现,直到1856年人们在德国杜塞尔多夫附近尼安德特山谷的一个山洞里又发现了尼安德特人的头骨和其他骨骼,这些骨头的主人才获得了“尼安德特人”的名称。相比起来,丹尼索瓦人发现的时间则远远晚于尼安德特人。2008年科研人员在西伯利亚南部阿尔泰山脉的丹尼索瓦洞穴中挖掘出了一具来自少年古人类的远端手指骨。斯万特·帕博及其团队[3]利用这具被发现的远端手指骨提取残留在里面的DNA,并对该古人类的基因组进行测序,覆盖率大约为1.9倍。结果表明,这个手指骨主人的DNA是独一无二的,这显然是一个既不同于尼安德特人,也不同于现代人的群体,于是将其命名为丹尼索瓦人,他们同样属于一个古人类群体,与尼安德特人有着共同的祖先,但却有着不同的种群(population)历史。当然,丹尼索瓦人与尼安德特人并不是两个完全独立的群体,事实上这两个群体之间也存在着基因交流。在斯万特·帕博2018年的研究中,他和他的团队获得了丹尼索瓦洞穴中的骨头碎片“丹尼索瓦11号”,并对其进行了基因组的测序。他们不仅比较了骨头碎片中的基因组与来自尼安德特人基因组或丹尼索瓦人基因组的衍生等位基因相匹配的DNA 片段的比例,还绘制了基因组中位点的分布图,发现丹尼索瓦11 号携带一个与阿尔泰尼安德特人基因组相匹配的等位基因,以及一个与丹尼索瓦3号基因组相匹配的不同等位基因[4]。这种结果就是由尼安德特人与丹尼索瓦人发生基因交流后产生的,更确切地说,她是尼安德特人母亲和丹尼索瓦人父亲的后代,也是目前为止发现的第一代尼安德特人-丹尼索瓦人后代。多方研究共同表明,无论是尼安德特人还是丹尼索瓦人,他们都不是一直处于孤立的状态,他们也与现代人的祖先存在着基因交流,现代人也依然延续着他们的基因。
1.2 古人类基因研究方法的开创与线粒体DNA的研究
在开展整个研究的过程中,存在各种各样的问题,有技术方面的,当然也有因为年代久远,导致提取到的DNA 存在人为或环境的污染、含量低等问题,为了让研究持续进行,斯万特·帕博不断革新技术,让研究得以从mtDNA 到核DNA 实现飞跃。
1.2.1 硅基质纯化法
聚合酶链式反应(PCR)的发展让研究已灭绝的物种和过去的种群成为可能,但它面临着两方面问题的考验。一方面是提取物往往含有抑制Taq DNA 聚合酶的成分,导致考古遗迹不能产生可扩增的DNA。另一方面则是聚合酶和微量当代DNA的污染可能产生误导的结果。为了更好地提取DNA,基于Boom 等[5]提出的核酸纯化方法,斯万特·帕博及其团队对该纯化方法进行修订,构建了硅基质纯化法。主要做了两方面的改进:a.减少处理过程中存在的包括DNA 的污染;
b.克服二氧化硅对DNA 双倍化造成的双倍污染。改善之后的方法能够高效提取残存在骨头中的DNA,并且考古提取物成分对PCR 的抑制作用也能够很好地被消除[6]。
1.2.2 利用氨基酸消旋确定内源性DNA
依靠PCR 和硅基质纯化法能够提取并扩增DNA[7],但是,仅有少数古代标本含有可扩增的古代DNA,而微量DNA污染造成的假阳性对古人类基因组学的研究构成了严重的威胁。为了解决这个问题,斯万特·帕博及其团队成功证明了天冬氨酸、丙氨酸和亮氨酸外消旋化的程度可以为评估古代组织样品是否含有内源性DNA提供参考。
除甘氨酸外,蛋白质中的所有氨基酸都能以两种光学异构体的形式存在,即D-和L-对映体,其中L-对映体专门用于蛋白质生物合成。氨基酸的外消旋化是指已有特定旋光性的氨基酸转变成另一种旋光性氨基酸的过程。一旦氨基酸从活性代谢过程中分离出来,L-氨基酸就会经历消旋化产生D-氨基酸,直到氨基酸的L-和D-对映体等量存在。斯万特·帕博及其团队将分析限定在符合许多真实性标准的9个案例中,并排除了人类遗骸(因为识别来自当代人的污染存在固有的困难)。他们还分析了17个样本,其中包括无法扩增出古代DNA序列的人类样本。结果发现,在D/LAsp 比值高于0.08 的样品中无法检索到古代DNA 序列,而所有比值低于0.08 的样品都产生了古代DNA 序列。这表明天冬氨酸的外消旋化程度可以评估古代组织样品是否含有内源性DNA。以前通过氨基酸的消旋作用,判断化石的年代和海岸线的变迁[8],现在,在斯万特·帕博开创性的引领下,可以通过某些氨基酸的外消旋化程度来确定待测样品中是否含有内源性DNA。
1.2.3 选择mtDNA作为研究切入
人体内的mtDNA,有以下几个特点:一是具有高拷贝数并且明显缺乏重组[9],二是具有高替代率[10],三是母系遗传[11]。鉴于这几种特点,帕博及其团队认为mtDNA 是一种对于研究和理解人类进化的有力工具。而且由于进化速度很快,可以用它来对进化过程进行高分辨率分析[10]。然而,几乎所有基于mtDNA 测序的人类进化研究都局限于控制区,而这个控制区占线粒体总DNA 不到7%[12]。这个控制区在生物学上也被称作D 环区,包含在非编码区的范畴内。为了研究进化谱系,科学家们又开创了“分子钟假说”,并且他们认为让这个分子钟假说成立的先决条件是对于任意给定的大分子蛋白质或DNA 序列在所有演化谱系中的演化速率近似恒定[13]。帕博及其团队在研究人类进化过程中无法避免地要以该假说为工具。而前面提到的控制区并没有在所有人类血统中以恒定的速率进化,但是人类的mtDNA 序列(不包括D 环)的进化速率大致相同,并且和黑猩猩mtDNA 的进化速率也没有显著差异[12],表明控制区对于研究人类进化并没有太大的意义。因此,为了不让分析局限于控制区,帕博及其团队决定对53 名不同来源的人类完整mtDNA 序列进行分析,进而描述人类的全球mtDNA多样性[12]。2004年,帕博及其团队对来自德国、俄罗斯和克罗地亚的4个尼安德特人化石的mtDNA 进行检索,发现这些个体携带了目前人类中尚未发现的mtDNA。通过与5个早期现代人进行比较发现,这5个早期现代人中没有一个含有这种mtDNA 序列。结合已有的mtDNA 数据,他们认为可以排除尼安德特人对早期现代人类的任何重大遗传贡献,但不排除较小贡献的可能性[14]。2008年,他们还采取高通量测序技术从38 000 岁的尼安德特人个体中重建完整的线粒体基因组序列,从中得出尼安德特人有效种群规模很小的结论[15]。2009年,帕博及其团队提出了一种古代DNA 序列检索方法,这个方法不仅有针对性,而且大大减少了样本破坏和测序需求,他们使用这种方法重建了5个尼安德特人的完整mtDNA基因组,除此之外,他们还做了对mtDNA 蛋白质进化的分析,结合这些数据他们最后发现尼安德特人长期有效种群规模小于现代人类和现存的类人猿[16]。
1.3 技术的发展使研究转向核基因组
由于mtDNA 仅是研究古人类基因组学的冰山一角,为了更加全面地研究人类进化与起源,在技术发展的帮助下,帕博及其团队逐渐把研究转向了尼安德特人的核基因组。454生命科学公司提供了一个全新的高通量基因检测系统。这个系统的优势在于可以在体外分离扩增并检测DNA,且一次性可以检测数百万碱基中所含的信息。基于这项技术,斯万特·帕博团队将研究重点放在了核基因组的检测中。由于这项系统每次能产生几十万个DNA 拷贝,还可以直接测序。避免了在PCR 中定向选择时引物错误造成的一些影响。最重要的是,其规避了细菌克隆的需要,因为细菌克隆会损失大量的模板分子。它进一步排除了PCR 中的模板竞争,因为每个分子都是单独扩增的[17]。这些新技术使核基因组分析的效率大大提高。
斯万特·帕博成功测出了尼安德特人的1 000 000个基因,并较为精确推算出尼安德特人与原始智人的分离时间,这使斯万特·帕博及他的团队信心大增。于是他提出了新的目标与计划,要得到尼安德特人基因序列草图。他在洁净的实验室添加了一种新的接头,这种接头包含尼安德特人特有的密钥序列,可以准确识别被确定为尼安德特人骨头提取液当中的基因序列[18]。这个装置可以被用来估计尼安德特人化石提取液中内源mtDNA 和污染人类mtDNA 的相对数量[19]以及由碱基对之间的氧化或者错配导致的基因序列错误,从而可以避免很多后续实验当中所产生的测序误差[20],同时,他改进了测序的文库技术,通过把最后一步实验用NaOH 处理改为用90°C 孵育处理,让最后一步的回收率与前面的步骤保持相近。
斯万特·帕博及他的团队使用了更新的测序方法,他们使用Ⅰllumina 基因组分析仪来分析DNA序列,这种分析仪是基于并行以及荧光的读取方法,来对数百万序列进行分析,同时可以使用可逆终止子进行迭代测序[21]。这种方法的优势在于解决了测序结束时总是会出现的碱基对错配情况(C-T的转变),从而使测序结果进一步精确。
1.4 尼安德特人的种群特点
1.4.1 高覆盖率的丹尼索瓦基因使种群分离时间被重新估算
斯万特·帕博团队在2008年的一块原始骨头上发现了高覆盖率的(>30 倍)丹尼索瓦人基因[22],不久后又在这个丹尼索瓦人居住过的洞中发现了一块人近端脚趾指骨,通过高通量DNA 测序发现,这块指骨上有高覆盖率的尼安德特人基因。这项研究成果不仅表明了尼安德特人的活动范围一直延伸到了西伯利亚的阿尔泰山,还说明了丹尼索瓦人和尼安德特人之间应该有基因交流。他们将该基因组与现有的低覆盖率尼安德特人基因组(包括来自高加索地区Mezmaiskaya 洞的一个基因组)以及25个高覆盖率的现今人类基因组进行了比较分析,斯万特·帕博团队估计尼安德特人和丹尼索瓦人之间的种群之间的分离(population segregation)是在38 万~47 万年前,现代人和尼安德特人/丹尼索瓦人之间的分离时间是在55 万~76万年前。
1.4.2 尼安德特人的遗传多样性较低
高通量测序在古人类基因组的逐渐推广使核基因组测序信息逐渐完善。许多尼安德特人的族群信息被斯万特·帕博团队分析出来。2014年,斯万特·帕博小组使用杂交方法从西班牙和克罗地亚的两个尼安德特人基因组的蛋白质编码部分富集DNA,并将这些序列与高覆盖率的西伯利亚尼安德特人的基因组一起进行分析。结果显示,尼安德特人的遗传多样性较低,不同族群之间相互隔离、封闭,发生较少的基因交流。斯万特·帕博小组随后发表了第三个高覆盖率的尼安德特人基因组[23],她是来自阿尔泰山Chagyrskaya洞的个体。据分析,她与来自欧亚大陆西部的尼安德特人的亲缘比来自西伯利亚南部的丹尼索瓦洞的尼安德特人亲缘更近。这与较为早期的尼安德特人灭绝和尼安德特人从欧亚大陆西部向东部迁徙的事实较为符合。上述研究结果表明,位于西伯利亚的尼安德特人族群较小,较分散,而居于欧洲的尼安德特人、阿尔泰山的丹尼索瓦人和古代现代人都居住于较大的部落当中。
2.1 抗病毒信号STAT2基因的渗入
2.1.1 STAT2对于抗感染的作用
STAT 家族由7个转录因子组成,每个转录因子包含7个结构和功能上保守的结构域:N端结构域(NTD)、卷曲螺旋结构域(CCD)、DNA 结合结构域(DBD)、接头结构域(LD)、Src同源2结构域(SH2D)、酪氨酸磷酸化位点(pY)和转录激活结构域(TAD)[24]。STAT2在其核心功能上基因具有高度保守性。
ⅠFNs 由3个主要的亚家族组成,分别为Ⅰ型、ⅠⅠ型和ⅠⅠⅠ型。Ⅰ型干扰素(ⅠFN-Ⅰ)由ⅠFN-β、ⅠFN-κ、ⅠFN-ω、ⅠFN-ε和ⅠFN-α等13种亚型组成;
ⅠⅠ型干扰素(ⅠFN-ⅠⅠ)由 单一的ⅠFNγ 组成;
ⅠⅠⅠ型干扰素(ⅠFN-ⅠⅠⅠ)由ⅠFN-λ1、ⅠFN-λ2、ⅠFN-λ3[25]和ⅠFN-λ4[26]组成。ⅠFNs 主要由浆细胞、树突状细胞产生,以回应特异性受体的刺激信号,这些受体位于细胞表面、细胞膜或细胞内[27],接受微生物产物或外来核酸的刺激[28]。
酪氨酸的磷酸化会导致两个相同或者不同的STAT 分子形成同源或异源二聚体(ⅠFN-Ⅰ诱导STAT蛋白酪氨酸残基磷酸化,通过SH2-pTyr相互作用介导STAT 发生平行二聚体构象的二聚化[29])。在ⅠFN-Ⅰ诱导信号的典型途径中,STAT1在Tyr701 处的磷酸化和STAT2 在Tyr690 处的磷酸化会诱导平行构象中的异质二聚体,并与ⅠRF9 相互作用最终形成ⅠSGF3复合物(图2)。ⅠSGF3复合物被转运到细胞核中,在那里含ⅠSGF3 的STAT 二聚体与基因启动子结合,特别是ⅠFN-Ⅰ刺激反应元件(ⅠSRE),它含有共识序列AGTTTCN2TTTCN,可以激活300 多个干扰素刺激基因(ⅠSG)[30]的转录,最终提高机体的抗感染能力。

Fig.2 The influence of ancient human gene infiltration on the anti-infection ability of different contemporary races图2 古人类基因渗入对当代不同人种抗感染力的影响
2.1.2 尼安德特人引入的STAT2单倍型
研究者对6个旧世界人群(比亚卡人(Biaka)、曼登卡人(Mandenka)、桑人(San)、汉人(Han Chinese)、法属巴斯克人(French Basque)、巴布亚新几内亚人(Papua New Guineans))的STAT2(包括所有的编码外显子)进行了重新测序,观察到存在一种仅限于非非洲人群的单倍型(N),并且它有相对较深的分支。该单倍型与尼安德特人共享衍生的SNP,在非非洲人中产生扩展的连锁不平衡(LD),并显示出与尼安德特人序列较高的同源性[31]。并且单倍型N 在巴布亚新几内亚以10 倍的频率被发现,表明STAT2成为美拉尼西亚人正向选择的基因之一。N单倍型在西亚人中延伸约130 kb,在一些东亚人和美拉尼西亚人中延伸至约260 kb,产生的LD 比在撒哈拉以南非洲人中观察到的强得多。从这些数据可以得出,现代人在欧亚大陆的祖先与尼安德特人进行基因交流时,引入了这一种在环境中十分有选择优势的免疫基因STAT2,并且这种基因的单倍型N在巴布亚新几内亚有着较高的频率。
2.2 OAS基因组区域与免疫
2020年,在遗传关联研究以及来自重症监护中易感性和死亡率的遗传学(genomics of mortality in critical care,GenOMⅠCC)联盟的数据支持下,斯万特·帕博团队研究发现,与感染严重急性呼吸综合征冠状病毒2(SARS-CoV-2)时需要重症监护有关的来自12 号染色体上某个区域的单倍型是从尼安德特人遗传而来的[32]。而该基因组区域包含3个基因,分别是OAS1、OAS2 和OAS3。这3个基因均位于12q24.1,并且都来自OAS家族。OAS家族蛋白由OAS1、OAS2、OAS3和OAS 样蛋白(OASL)组成。OAS1~3 蛋白具有明显的同源性,它们仅在OAS 单元的数量上存在差异[33]。作为抗病毒过程中的重要物质,它们会在干扰素的配合下,一起将病毒消灭。
干扰素是最强大的抗病毒细胞因子,可以诱导像OAS1、OAS2和OAS3这些能够介导抗病毒效应的基因产生相应的反应。前面提到的3种蛋白质均可以通过核糖核酸酶L(RNase L)依赖途径对入侵的病毒产生抵抗作用。当病毒感染宿主细胞后,病毒的双链RNA(dsRNA)会刺激OAS1、OAS2和OAS3,在干扰素的诱导激活下这些基因会表达,产生寡腺苷酸合成酶,但是此时产生的酶仍以酶原的形式存在[34],所以还需要dsRNA 的激活。寡腺苷酸合成酶在被激活以后会催化2"-5"寡核苷酸的合成,然后激活核糖核酸酶RNase L。激活的RNase L会降解细胞内的dsRNA(图2),同时它也会激活细胞内其他抗病毒机制。例如,MDA5 和RⅠG-Ⅰ识别裂解的病毒RNA后,会促进ⅠRF3 和ⅠRF7 的激活,进而引起一系列其他反应[33]。除了RNase L 依赖途径,OAS3 还可以通过另一种途径发挥作用,即非RNase L依赖途径。许多研究都已经表明OAS 家族蛋白在对抗RSV、流感病毒、登革热病毒、HⅠV 等病毒的过程中发挥了重要的作用[34],当然这其中也包括冠状病毒。OAS1~3所对抗的病毒范围相对来说比较广泛,可以更多地挖掘它们在抗感染方面的潜能。
2.3 TOLL样受体的反复渗入
根据研究表明,在10 种人类Toll 样受体中,有3 种在4 号染色体上:TLR10、TLR6、TLR1。这3 种能表现出尼安德特人的高渗透率,并且这3个TLR 基因发挥着先天免疫的关键作用[35-36]。先天免疫系统提供抵御病原体的第一道防线,参与微生物的早期检测以及适应性免疫反应的激活。大多数TLR 的刺激导致Th1 而不是Th2 分化,Th1 细胞产生干扰素-g(ⅠFN-g)并介导细菌和病毒感染的消除,而产生ⅠL-4 和ⅠL-13 的Th2 细胞参与对抗蠕虫感染的反应。细菌一些独特的细胞壁成分(如LPS)会刺激免疫细胞并作为病原相关分子模式(PAMP),被单个TLR 识别。LPS,也被称为内毒素,通常是细胞壁成分中最有效的免疫刺激剂[37]。在革兰氏阳性和革兰氏阴性细菌中都存在的脂蛋白和肽聚糖(PG)也是有效的免疫刺激剂。TLR2在检测革兰氏阳性菌中起主要作用,并参与识别各种微生物成分,包括LTA、脂蛋白和PG。TLR2 与TLR1、TLR6在物理和功能上相互作用,它们似乎参与识别脂蛋白脂部分的细微变化[36]。白细胞中TLR6、TLR1和TLR10的表达显著增加。而在现代人中,幽门螺杆菌的血清阳性率降低和对过敏的易感性增加[38]也能说明TLR基因在免疫当中的重要作用。综上所述,渗入的等位基因可能增强先天免疫监视和对某些病原体的反应性,但这也可能会增加对非病原体过敏原的超敏性,从而导致当今人类出现过敏性疾病。
2.4 病毒互作蛋白(VIP)基因的交流
在最近的10 万年当中,尼安德特人和古现代人至少杂交过两次,在杂交的过程中不可避免地会发生核基因组的交流,两类人种当中都会存在对方相应的等位基因,在自然的正向选择当中优势基因会发生保留,并以相对较高的基因频率存在于人群当中。然而这些基因交流,其实也就相当于每个物种暴露于对方物种提供的新病毒的环境下,并且彼此提供对这些病毒抗性的适应性等位基因。其中的一类就是病毒互作蛋白(VⅠP)。STAR 分析表明,相比于和DNA 病毒相互作用的VⅠP,和RNA 病毒特异性相互作用的VⅠP在现代欧洲基因组中特别丰富[39]。现在已经发现了20 种已知能与至少10个VⅠP相互作用的人类病毒。相关病毒倾向使用类似的宿主VⅠP[40],比如与HⅠV相互作用的VⅠP也可能与其他慢病毒相互作用。4 534个人类VⅠPs 与非VⅠPs 相比,VⅠPs 表现出:a.非同义多态性与同义多态性的平均比例较低;
b.罕见的、可能有害的多态性比例较高;
c. 可能有害的分离变体的密度较高[41]。VⅠP 还在保守基因组片段密度较高的区域被发现[42],同时它在人类蛋白质-蛋白质相互作用网络当中比非VⅠP 有更多相互作用的蛋白质伙伴[43]。并且研究表明,因为自然正向选择使尼安德特人具有高度VⅠPs 富集,并且在VⅠPs 处重叠的片段比与非VⅠPs重叠的片段更长,频率更高[39]。
总的来说,关于病毒适应性的引入情况,不同地区的引入率不同或许与不同地区自然环境下病毒的正向选择有关。原始现代人和尼安德特人之间的基因交流允许特定的病毒VⅠPs 进行跨物种传播,使双方物种都具有对特定病毒的抵抗性。在欧洲而不是在东亚的RNA VⅠPs 上的强烈富集表明,这种差异是由亚洲人和欧洲人分离后的脉冲式杂交产生的[44]。
2020年,重度新型冠状病毒肺炎(COVⅠD-19)全基因组关联分析(genome-wide association study,GWAS)组在意大利和西班牙征集志愿者,对符合标准的志愿者进行基因分型和分析。通过做荟萃分析,他们发现有2个位点与COVⅠD-19 诱导的呼吸衰竭相关。这两个位点是3p21.31 和9q34.2。其中3p21.31 位点里包含6个与COVⅠD-19 有关的基因。这6个 基 因 是SLC6A20、 LZTFL1、 CCR9、FYCO1、CXCR6 和XCR1[45],这6个基因相关的基本信息和发挥作用的机制见表1。同年,基于这项研究[45],斯万特·帕博发现,能加重COVⅠD-19的最相关遗传变异均处于高连锁不平衡状态,他开始调查这个位点与古人类有无关系。他还参考了由COVⅠD-19 宿主遗传学倡议(The COVⅠD-19 Host Genetics Ⅰnitiative)发起的研究[46],分析了1 000个基因组项目中该基因组区域的所有5 008个单倍型,最后发现遗留在现代人体内的这个位点其实来源于尼安德特人。因此尼安德特人单倍型可能是一些人群中患重症COVⅠD-19的重要因素。被留下基因印迹的人里,南亚携带者频率最高,欧洲和美国次之,东亚最低。这个发现或许可以解释为什么中国人患COVⅠD-19 以后发展为重症的比例很低[47]。

Table 1 Basic information of ancient introgressive genes and their mechanism表1 古代渗入的基本信息及发挥作用的机制
与重症COVⅠD-19 有关的这6个基因中有3个基因编码的蛋白质都是趋化因子的受体,分别是CXCR6、XCR1 和CCR9。这3个受体和它们相应的配体作为免疫方面信号通路的一部分,介导细胞的趋化性,在机体对抗病原体的过程中发挥着重要作用[48-50]。其中CCR9 作为β 趋化因子受体家族的一员,目前被证明可以调节肺嗜酸性粒细胞的聚集[51],这可能也与COVⅠD-19 有一定相关性。另外3个基因虽然不与趋化因子直接相关,但也同样在免疫过程中发挥作用。作为一种转运体,SLC6A20 主要定位于质膜上,主要转运的是脯氨酸[52]。SLC6家族中的转运体需要Collectrin(一种ACE2的同源物,在肾脏氨基酸及Na+转运以及胰岛素的分泌等方面具有重要作用)或者血管紧张素转换酶2(ACE2,SARS-CoV-2 细胞表面受体)才能到达细胞表面[53]。SLC6A20 在功能上与ACE2 相互作用,因此研究人员将它视为与COVⅠD-19重症有关的候选基因[45]。同时也有研究表明,SLC6A20表达增加可能导致ACE2蛋白水平增加和病毒摄取增加[51]。LZTFL1 在纤毛细胞内高度表达,它能够调节气道纤毛的发生和功能,这对气道清除病毒起着重要作用。以COVⅠD-19为例,如果该基因表达减弱,那么纤毛发挥的作用可能就会受到影响,气道清除病毒的效率会下降,进而刺激更多SARS-CoV-2在肺部积累,加重病情[54]。除此之外,在病毒感染的过程中,上皮-间充质转化(epithelial-mesenchymal transition,EMT) 会导致SARS-CoV-2 的两种细胞受体减少:ACE2 和跨膜蛋白酶丝氨酸2(TMPRSS2)。研究表明,LZTFL1水平升高抑制EMT,而LZTFL1 水平降低促进EMT[55]。所以当LZTFL1 表达升高时,EMT 会被抑制,那么前面提到的两种受体就无法明显减少,抑制SARS-CoV-2 感染的效率也会大大降低。FYCO1 编码Rab7 衔接蛋白,它能够与LC3、PtdⅠns(3)P和Rab7结合,也能与自噬体外膜结合,所以研究人员又将它称为自噬膜和微管以及末端定向分子马达之间的适配器[56]。同时,FYCO1是连接ER衍生的双膜囊泡(冠状病毒的主要复制位点)与微管网络的关键介体[57]。
以上提到的6个基因在不同程度上都与免疫建立了一定的联系,抗击SARS-CoV-2的过程也与免疫密切相关。目前有许多的研究都在探究它们与COVⅠD-19 的关系,目前尚未阐明这6个基因具体是如何导致重症COVⅠD-19的,但可以明确的是该基因簇是COVⅠD-19 患者的遗传易感位点[45],它们可以成为COVⅠD-19药物研发的切入点。
截至2017年,2型糖尿病患者超过4.15亿,是全球发病率和死亡率的主要原因[58]。目前研究人员们已发现117 种可能导致2 型糖尿病的基因,而SLC16A11基因就属于其中一个。近年来越来越多的研究表明,SLC16A11 基因与2 型糖尿病密切相关,与前面提到的3号和12号染色体上的两个基因簇一样,这个位于17 号染色体的基因同样来自于尼安德特人[59]。SLC16A11 基因隶属于SLC16 基因家族,这个家族也被称作单羧酸转运体家族。SLC16A11 基因作为其中一员,编码蛋白质MCT11,它在肝脏、皮肤、肺、卵巢、乳腺、胰腺等部位中表达[60]。科学家们对SLC16 家族做了分类,其中第一类成员的功能是通过质子耦合机制运输简单的单羧酸,如乳酸、丙酮酸和酮体。SLC16A1、SLC16A3、SLC116A7 和SLC16A8 都归为第一类成员,在最近的研究中,也发现SLC16A11是一种质子耦合单羧酸转运体,所以它可以发挥与其他4位成员相似的作用。人体细胞内存在着两种伴侣蛋白,分别是basigin 和embigin,它们都由一个包含保守谷氨酸残基的单个跨膜结构域、一个短的细胞内C端和一个大的糖基化细胞外结构域构成,它们需要在SLC16家族第一类成员的帮助下才能到达质膜,才能在高尔基体中积累。如果它们的活性降低,可能会导致乳酸流出,进而影响糖酵解速率[58]。在小鼠体内被敲除SLC16A11基因,再将变异的SLC16A11基因编码的异常蛋白转入基因敲除小鼠体内后,研究人员发现,小鼠体内发生了脂蛋白的上调,这导致了更多的甘油三酯积累和胰岛素抵抗,这说明SLC16A11突变会影响脂质代谢的异常功能。在ⅠⅠ型糖尿病患者中开展的研究也同样发现甘油三酯在肝组织中堆积,而这种堆积与人类的胰岛素抵抗有关,所以研究人员推测SLC16A11变异体可能通过调节脂质代谢来增加糖尿病风险,但如何直接导致代谢紊乱尚不清楚[61]。关于SLC16A11基因与2型糖尿病的研究还在继续,无论具体的机制如何,目前的研究都能为科研人员深入研究指明方向,对药物开发也提供了一定的指导,比如弥补因为SLC16A11 突变导致的功能缺陷。
随着现代人类迁出非洲,他们遇到了许多新的环境条件,包括更高的极端温度、不同的病原体和更高的海拔。这些不同的环境很可能充当了自然选择的有效途径,并促使人们适应当地的环境。最著名的例子之一是西藏人适应青藏高原的缺氧环境[62]。在高原缺氧环境下选择的最强信号来自EPAS1,一种参与缺氧反应的转录因子[63]。EPAS1也称为缺氧诱导因子2α(HⅠF-2α)。HⅠF 转录因子家族由两个亚基组成,其中3个交替的α 亚基(HⅠF-1α、HⅠF2α/EPAS1、HⅠF-3α)与由ARNT 或ARNT2 编码的β 亚基二聚化。HⅠF-1α 和EPAS1 各自作用于一组独特的调节靶点,EPAS1的表达谱包括成人和胎儿肺、胎盘和血管内皮细胞。EPAS1中的蛋白质稳定突变与红细胞增多症有关[64],表明EPAS1与红细胞生成的调节之间存在联系。但是在青藏高原EPAS1 基因高表达的个体却与低红细胞数量相关。红细胞产生是对缺氧应激的常见反应,可能是EPAS1“西藏等位基因”的携带者能够在高海拔地区维持足够的组织氧合,而无需增加红细胞水平。关于EPAS1 基因具体在缺氧呼吸时发挥作用的分子机制还有待考究。
斯万特·帕博团队对尼安德特人和丹尼索瓦人的不断探索与发现,不仅开创了一个古人类基因组学的新学科,而且从生物、地理、历史角度完善了“走出非洲”这一人类进化学说。他的发现也给予古生物学很好的发展思路和前进方向,不仅加深了研究人员对古生物种群特征的了解,而且为他们的进一步溯源工作提供了良好的基础。同时该项研究紧紧跟随时代最新的分子生物学技术,比如优化回收DNA 的手段、高通量测序的普及、完善构建基因文库等技术。这些新的方法、新的技术无疑为进一步开展人类基因组溯源提供了更好的途径。斯万特·帕博的工作为当代遗传学疾病研究、流行病学调查、免疫学等提供了一个新的切入点。面对全球性的疾病爆发,通过人类基因追溯工作,更能洞悉不同地区人种的基因差异性,有利于合理分配不同地区的医疗资源,以及公共卫生工作的进一步开展。相信在不久的将来,人们能通过对古人类基因组的研究,更好地理解现代人基因的来源与去向,攻克更多的疑难杂症,真正读懂老祖宗留下的信息,更好地去造福后代。
猜你喜欢 尼安德特人丹尼 最后的尼安德特人小哥白尼(趣味科学)(2022年4期)2022-06-30不吃蔬菜的因纽特人 北极圈幼儿100(2021年10期)2021-04-25尼安德特人的鹰骨饰物大自然探索(2019年9期)2019-12-13乌龟丹尼快乐作文·高年级(2017年9期)2018-01-15原谅那个保罗分忧(2015年6期)2015-06-09关于尼安德特人你所不知的20件事奥秘(2014年7期)2014-09-10世界末日故事会(2006年3期)2006-01-12本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0611/610034.html