【www.zhangdahai.com--其他范文】
任艳丽, 蔡文红, 陈庶伟, 胡燕华, 杨长仪
先天性中枢性低通气综合征(congenital central hypoventilation syndrome,CCHS)是一类罕见的常染色体显性遗传病,主要表现为因化学感受器对高碳酸血症和低氧血症反应低下而引起的肺泡通气不足[1]。于20世纪70年代首次报道,活产儿发病率约为1/200 000[2],表现为非肺、神经系统或代谢性疾病所引起的呼吸暂停以及二氧化碳潴留,常于睡眠状态下出现严重的低通气,严重者于清醒状态下也会出现低通气表现,伴或不伴有自主神经系统失调疾病,如消化系统发育异常(先天性巨结肠)、神经嵴源性肿瘤等。93%~100%的CCHS病例与PHOX2B基因突变有关,也有少数与其他基因相关,如RET、GDNF、EDN3、BDNF和ASCL1等[3]。PHOX2B存在2类突变,一是聚丙氨酸重复扩展突变(polyalanine repeat expansion mutations,PARMs)(约占90%);
二是非PARMs(nonpolyalanine repeat expansion mutations,NPARMs)(约占10%)。诊断依赖于临床表现及基因诊断,因对疾病认识不足,易误诊或漏诊,尤其是以呼吸暂停为常见症状的早产儿,更易误诊。本研究报道1例CCHS早产儿并进行文献复习,为临床早期诊断和及时干预提供依据。
1.1 一般资料 患儿,女,33+1周,因出生后呼吸困难收治住院。胎膜早破15 h,羊水清,无窒息,出生体质量1 090 g,其母产前2周促胎肺成熟治疗1个疗程。产前2个月,母亲因诊断为“亚临床甲状腺功能减退”,以雷替斯(左甲状腺素片)替代治疗;
产前20 d,彩超提示胎儿双顶径、腹围小于相应孕周2个标准差;
产前9 d,彩超提示羊水过多(羊水指数42.7 cm)。
1.2 各系统表现
1.2.1 呼吸系统 患儿出生后即出现呼吸困难,无法离氧,入院后予无创通气-双水平气道正压通气(bilevel positive airway pressure, BiPAP)、特治星抗感染、枸橼酸咖啡因兴奋呼吸中枢、静脉营养支持等治疗。出生3 d,因反复呼吸暂停予气管插管及同步间歇指令通气(synchronized intermittent mandatory ventilation, SIMV);
出生4 d,转为无创通气-经鼻间歇正压通气(nasal intermittent positive pressure ventilation, NIPPV);
出生7 d,转为BiPAP模式,8 h后动态监测二氧化碳分压,提示高碳酸血症(79 mmHg,1 mmHg=133.3 Pa),调整为NIPPV模式呼吸支持;
出生13 d,出现反复呼吸暂停伴二氧化碳潴留(65 mmHg),予气管插管SIMV模式呼吸支持;
出生15 d,BiPAP下反复呼吸暂停并伴二氧化碳潴留(79 mmHg),再次气管插管高频振荡通气(high frequency oscillatory ventilation, HFOV)模式呼吸支持。
1.2.2 消化系统 出生1 d开始微量胃肠道喂养,出生4 d安敏健/母乳1 mL/3 h下出现消化道出血,出生13 d奶量4 mL/3 h下出现腹胀,出生19 d再次胃肠道喂养(氨基酸奶),出生25 d奶量12 mL/3 h下腹胀明显。患儿大便排出异常,无自主排便,多次通便处理亦无大便排出,仅于生后6、9、12 d通便后排出少量墨绿色大便。
1.3 辅助检查 出生当天及出生后4 d胸片提示肺部纹理增多。出生后4、6、7 d腹部平片提示肠管扩张,形态欠规则,略呈管型。出生后13 d腹部立位片提示低位肠梗阻。出生后25 d腹部彩超提示肠蠕动减弱,肠管扩张,肠腔内存多量内容物;
心脏彩超提示卵圆孔未闭;
颅脑彩超提示双侧室管膜下出血,双侧脑室增宽(左侧0.5 cm,右侧0.6 cm)。住院期间动态血常规及C反应蛋白大致正常,动态生化指标提示低蛋白血症,余正常。血、痰、大便培养均为阴性,真菌葡聚糖实验阴性。
1.4 转归 因患儿生后反复呼吸暂停伴二氧化碳潴留,需有创呼吸机支持,反复腹胀,无法建立肠道内营养,于出生后26 d放弃治疗后死亡。
1.5 分子遗传学分析 征得患儿家属同意后,采集患儿外周血样本送武汉康圣达医学检验所行全外显子基因分析。根据患儿基因分析结果,征得家属同意,采集患儿父母外周血样本进行家系分析。
1.6 文献检索策略 以“先天性中枢性通气综合征”“婴儿”“PHOX2B基因突变”为检索词在中国知网、万方数据库、维普数据库和PubMed等数据库进行检索,检索时间为建库至2020年7月31日,检索语言为中文和英文。排除指南、共识、综述、动物实验以及非新生儿病例的文献。病例纳入标准:具备完整的临床资料,经分子遗传学检查确诊。对纳入病例的临床表现、分子遗传学检查结果、治疗及随访情况进行分析总结。好转定义为能成功过渡至家庭通气支持。
1.7 统计学处理 汇总文献病例及本病例,按基因检测结果分为PARMs和NPARMs组,采用χ2检验比较两组患儿的临床表现、治疗和预后情况。针对PARMs组,采用χ2检验比较不同数目丙氨酸扩增组患儿的临床表现、治疗以及预后情况,当20%以上理论频数<5时,采用Fisher确切概率检验。P<0.05为差别有统计学意义。
1.8 结果
1.8.1 分子遗传学分析 该患儿二代测序(高通量测序)结果提示,在PHOX2B基因第3外显子,染色体位置:chr4:41748329发现一处杂合突变(NM_003924:c.440 A>G),根据美国医学遗传学与基因组学学会指南,该突变分级评定为致病突变。一代验证提示父亲和母亲均为野生型(图1)。患儿在PHOX2B基因上发现的杂合突变,经家系验证考虑为新发突变(但不排除父亲或母亲存在生殖细胞突变嵌合体可能)。该例患儿按基因分型属于NPARMs。

图1 一代测序家系验证图(箭头表示突变位点)Fig.1 Family verification map of the first generation sequencing (arrow indicates mutation site)
1.8.2 文献检索结果及分析 共检索文献87篇(中文26篇,英文61篇);
排除综述17篇(中文9篇,英文8篇),非新生儿病例报道5篇(均为中文)以及其他非病例报道17篇(均为英文),符合要求的文献48篇(中文12篇,含病例22例;
英文36篇,含病例91例),其中33篇文献合计67例(中文14例[4-10],英文53例[3,11-35])有详细资料(图2)。对本病例及上述67例患儿的临床表现、分子遗传学以及治疗预后进行分析(表1,2)。由于为文献回顾,并非每个病例均含有所有研究指标,部分数据存在缺失情况。

图2 文献检索及病例筛选流程图Fig.2 Flow chart of literature retrieval and case screening
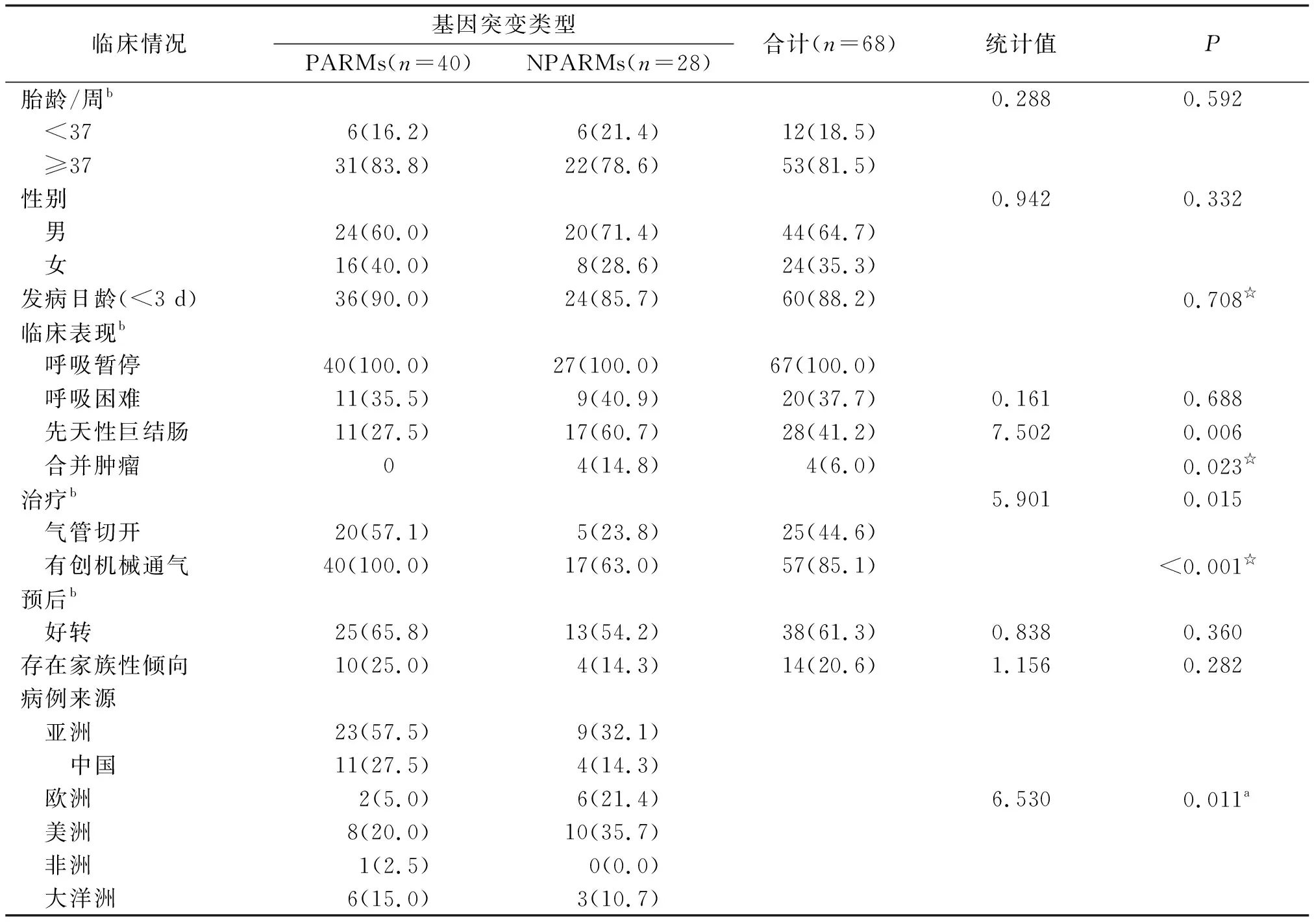
表1 基因检测及临床情况Tab.1 Gene detection and clinical situation

表2 不同聚丙氨酸扩展次数基因型资料比较Tab.2 Comparison of genotypes with different alanine expansion numbers
1.8.2.1 临床特点 68例中,足月儿53例(81.5%,53/65),男童44例(64.7%),于出生后3 d内发病60例(88.2%)。主要表现为呼吸暂停(100%,67/67),需要有创通气支持(83.8%,57/67);
并发消化系统疾病28例(41.2% ,28/68),合并肿瘤4例(6.0%,4/67),20.6%(14/68)的病例存在家族性因素。
1.8.2.2 不同基因型及临床表现比较 PARMs 40例,基因型分别为20/24(2例)、20/25(11例)、20/26(12例)、20/27(13例)、20/29(1例)和20/31(1例)。NPARMs 28例,突变方式分别为替换(12例)、缺失(10例)、重复(4例)、插入(2例)。NPARMs组发生先天性巨结肠和肿瘤的概率高于PARMs组(60.7%vs27.5%;
14.8%vs0,P<0.05),NPARMs组对有创通气和气管切开的需求低于PARMs组(63.0%vs100%;
23.8%vs57.1%,P<0.05)。丙氨酸重复次数多者,以男童居多(男女比例12∶3);
重复次数少者,发生先天性巨结肠较少(2/25vs9/15,P<0.05)、预后较好(21/25 & 4/13,P<0.05)、更有家族倾向性(10/25 & 0/15,P<0.05)。根据文献报道,基因型存在地域差别(χ2=6.530,P<0.05),亚洲的病例多为PARMs(69.7%),而欧洲和美洲多为NPARMs(64.0%)。
CCHS是一类罕见的以肺泡低通气为主要表现的常染色体显性遗传病[36],多于新生儿期发作[1]。目前全球已报道1 000余例,发病率无性别差异[3]。自CCHS被发现和报道以来,陆续有迟发性的病例报道,提示未经治疗者因长期肺泡低通气、严重缺氧而引起肺动脉高压、右心功能不全以及神经系统疾病不良预后[37]。近年来,越来越多的病例报道提示,CCHS患儿如能早期诊断和干预,大多数生存期可延长,并获得良好的生活质量。所以临床医师应提高对该疾病的认识,做到早诊早治。
CCHS的临床表现主要为反复发作的呼吸暂停伴二氧化碳潴留,但由于中枢对低氧血症以及高碳酸血症缺乏反馈机制,患儿可能并不出现呼吸节律增快,可伴发消化系统发育异常、神经嵴源性肿瘤及其他相关的自主神经改变,如眼部异常、食管运动功能障碍、便秘、感音神经性耳聋、心律失常、体位性低血压和神经症状(癫痫发作、发育迟缓)等[3]。此外,研究指出,>2岁的儿童和青少年患者存在特殊面容,主要是PARMs病例,通过5个变量来表征面容(上唇高度、眼距、上面部高度、鼻尖突出情况和嘴唇特征)可正确预测86%的CCHS病例和82%的对照病例[38]。本研究中患儿的临床特点为:男童、足月儿多见,发病时间多在出生后3 d内;
临床表现主要为呼吸暂停及二氧化碳潴留(100%),多数需有创机械通气(85.1%),部分需气管切开(44.6%),常合并先天性巨结肠(41.2%)和肿瘤(6.0%)。
CCHS主要与PHOX2B基因突变有关,临床表现与基因突变类型有一定相关性。PHOX2B是第4对常染色体上的配对同源盒基因,编码由314个氨基酸组成的转录因子,参与自主神经系统的分化与成熟[39],并调节多种基因的表达。PHOX2B存在2类突变,即PARMs和NPARMs。在正常个体中,PHOX2B外显子3的等位基因存在一段包含20个丙氨酸的重复片段(基因型20/20),而突变的等位基因丙氨酸重复次数为24~33(基因型20/24~20/33),此类突变即为PARMs。丙氨酸重复次数增加会加重疾病的严重程度,需呼吸机支持,并与较低的发病年龄相关[3]。NPARMs引起错义、无义、移码或终止密码子改变,此类基因突变病情常较重,需持续呼吸支持,合并先天性巨结肠以及神经嵴源性肿瘤等的风险高[40]。也有文献报道,NPARMs组在呼吸支持方面的要求可能低于PARMs组,仅表现为轻症的呼吸系统表型[30]。本研究结果显示,NPARMs组更多合并先天性巨结肠和肿瘤,呼吸系统表型较轻;
与丙氨酸重复次数多者相比,重复次数少的患儿合并先天性巨结肠较少,预后较好。提示该病存在一定的基因-表型相关性,基因检测有助于疾病的早期诊疗和预后评估。鉴于该病存在遗传倾向,美国胸科协会于2010年发表的CCHS管理声明建议[1]:针对曾生育该类患儿的父母予以基因检测进行家系分析,以评估未来子女发生疾病的风险,并提供遗传咨询;
通过基因检测进行产前诊断,预测后期发生肿瘤的风险,指导进一步的医学干预。由于PHOX2B基因突变类型各异,文献[41-42]推荐,针对不同的基因突变采用不同的方法进行检测:PHOX2B基因片段分析以筛查PARMs;
PHOX2B基因测序分析以明确NPARMs;
多重连接探针扩增技术检测PHOX2B外显子或者全基因的缺失/重复;
联合前两种检测方法筛查嵌合体。
针对CCHS的诊断,2020年发表的欧洲指南给出疾病诊断思路[43]:对于可疑低通气综合征的患儿,采用多导睡眠仪进行监测,如果存在睡眠相关的低通气则进一步行基因检测,否则应在排除感染、心肺疾患、神经系统疾病以及代谢性疾病等因素引起的呼吸暂停后行基因检测明确。确诊CCHS后,需注意排查是否合并先天性巨结肠、肿瘤以及其他自主神经功能紊乱等疾病。本研究报道的病例为早产、极低出生体质量儿,通过基因检测证实突变类型为NPARMs(基因替换)。该患儿临床表现与基因突变相符,证实基因-表型的相关性。既往文献报道的病例中,早产儿比例极低,提示可能因患儿为早产儿而忽略存在CCHS的可能性,在发病早期未进行基因诊断,从而导致误诊或漏诊。
目前CCHS尚无有效的治疗药物,需终身呼吸支持,主要治疗目标为维持最佳的通气(二氧化碳分压为35~45 mmHg)及氧合[43],根据患者年龄、发病年龄、呼吸机依赖的持续时间等,临床采用的治疗方式有:气管切开并正压通气、鼻罩或面罩的正压通气、负压通气或是膈肌起搏等。美国胸科协会2010年发表的CCHS管理声明建议[1]:一经确诊,在新生儿期或是婴儿期即可考虑行气管切开术。气管切开虽有利于长期呼吸支持,但也增加了感染的机会,且不利于患儿发声及语言交流,所以近年来开始尝试无创通气。国内已有3月龄患儿成功转为无创通气的文献报道[44]。考虑更好的气体交换以及神经系统发育,TRANG等[43]仍主张早期使用气管切开及正压通气支持,逐步过渡至无创通气(5~8岁),以避免发生神经系统不良预后以及面部发育不全。面罩正压通气的使用要求能够配合、呼吸道正常且仅在睡眠期间需要呼吸支持者,是仅需夜间通气的迟发型患者的首选。膈肌起搏可用于通气时间较长患者的日间呼吸支持。负压通气目前临床使用经验尚少。
综上所述,CCHS新生儿以呼吸暂停和二氧化碳潴留为主要临床表现,基因检测有助于明确诊断、预测临床表现及评估预后。对于CCHS的病例需要早期明确诊断和正确处理,预防和避免高碳酸血症、低氧血症所引起的不良预后。该病多在新生儿期发病,新生儿期如出现反复呼吸暂停及二氧化碳潴留,尤其是以呼吸暂停为常见临床表现的早产儿,需借助基因检测早期明确诊断,并注意排查相关并发症。
猜你喜欢 基因突变结肠先天性 精细化护理在先天性心脏病患儿围术期中的应用中国典型病例大全(2022年13期)2022-05-10胎儿心脏超声检查在先天性心脏病检测中的应用中国典型病例大全(2022年7期)2022-04-22携带线粒体12S rRNA基因突变的新生儿母系家族史分析中国听力语言康复科学杂志(2021年6期)2021-12-21左半结肠一期切除术探讨中国典型病例大全(2021年1期)2021-03-30久用泻药要警惕结肠黑变病家庭医学·下半月(2017年11期)2017-12-20“基因突变和基因重组”复习导航中学生理科应试(2017年6期)2017-09-27一例猫巨结肠症的诊疗中国动物保健(2015年4期)2015-10-21说说小儿先天性髋关节脱位为了孩子(孕0~3岁)(2001年3期)2001-06-13先天性畸形:矫治的最佳年龄祝您健康(1985年6期)1985-12-30本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0425/589081.html