【www.zhangdahai.com--其他范文】
徐斯然,吴奇,卢帮安,唐堂,张佳楠,*,胡劲松,*
1郑州市先进能源催化功能材料制备技术重点实验室,郑州大学材料科学与工程学院,郑州 450001
2北京分子科学国家实验室,分子纳米结构与纳米技术重点实验室,中国科学院化学研究所,北京 100190
3污染物分析与回收利用技术湖北省重点实验室,湖北师范大学化学化工学院,湖北 黄石 435002
能源短缺和环境污染加剧,迫切需要探索清洁能源技术,有效缓解化石燃料的需求压力1,2。氢能以其能量密度高、资源丰富等特点,成为目前最有希望替代化石燃料的清洁能源1,3。目前电力占全球总能源消耗的近20%,预计未来将超过石油和煤炭,2040年将超过30%。日趋增长的电力需求是可再生能源技术发展的主要驱动力,预计2040年三分之二的电力将会来自可再生资源4,5。在寻求零碳排放的过程中,电解水作为能够有效利用风能、水能或太阳能等可再生能源以可持续的方式转化为氢的最有潜力的途径之一,引起了广泛的关注6,7。然而,在水电解过程中,过电位的存在会造成能量损失8,析氢反应(HER)的效率在很大程度上取决于电催化剂的选择。因此,选择合适的催化剂可以有效地降低过电位,从而实现高效产氢。
然而,不同催化环境决定了催化剂的材料选择、设计原理和催化机制。根据所使用的电解质环境的不同,在酸性、中性或碱性条件下进行电解的催化剂性质各异。酸性环境会导致普通金属的严重腐蚀,因此需要使用稀缺的Ir、Pt等贵金属材料制备电极,从而导致高昂的资本成本,而且酸性高腐蚀环境大大削减了质子交换膜(PEM)电解槽器件以及电催化剂的寿命,一定程度上局限了酸性环境催化剂大规模应用及工业化发展9,10。相比之下,碱性介质下的电解水将催化剂的选择范围扩大至地球丰富的各类过渡金属材料,但通常工作效率较低,需要更大的器件,从而导致更高的成本。PEM电解槽的优点是高纯度的产氢能力,能够在高压环境下工作,以及能够快速储存氢燃料。与碱性电解槽相比,PEM固体聚合物膜作为一种优良的氧化还原反应屏障和气体分离屏障具有很高的反应潜力,赋予电解槽较高的质子电导率和氢不渗透性11,12。然而,碱性阴离子交换膜(AEM)的研发是碱性环境电解槽发展的一大进步。
便携式AEM电解槽系统完美地沿袭前期碱性电解槽系统低成本和PEM电解槽耐高压、产氢纯度高、效率高以及安全性高的优势的同时,还不需要高腐蚀性的液体电解质;
允许使用不昂贵的聚合物膜(如四元化聚合物基膜)替代高成本的Nafion基膜;
允许在小规模的电解水体系中高效工作13-15。因此,可以对低成本的AEM电解槽进行制造,并有助于发展稳定的产氢能力,增加耐久性,并提高固定式、便携式和运输式发电系统的能源效率16,17。然而,由于碱性HER的多反应步骤和水解离动力学,析氧反应(OER)的缓慢四电子转移动力学,制备高效电解水催化剂仍存在挑战。目前就如何精准控制催化剂特征活性位点仍是模糊不清的,因此,深入了解HER/OER的反应机理,进而设计高效的电化学水裂解催化剂至关重要。
面对工业化发展需求,开发能大规模制备、能在高电流密度下长时间高效工作、以及可承受压力和温度等工业相关条件下具有良好性能的电催化剂是至关重要的。在过去的十年中,越来越多的高效催化剂被开发设计,但大多数催化剂通常是在实验室条件下进行研究的(电流密度为1-100 mA·cm-2),有关电解水的研究普遍集中在基础材料的催化动力学上,只有少数催化剂能够处理工业相关的电流密度。而往往低电流密度下的催化剂并不能有效解决大电流密度下复杂的界面关系,气泡动力学以及欧姆降、温度、高压等问题4,8,18,19。然而,近年来,关于设计大电流密度下的工业电解水催化剂(电流密度> 1000 mA·cm-2)的研究工作层出不穷,然而鲜少有涉及到工业电解槽催化电极构建、膜组件设计以及器件组装等电池设计层面。
在此,本文着重对近年来碱性介质中电解水过程所涉及HER/OER目前公认的机理的解释与推论进行总结和讨论。随后,描述了目前电解水催化剂的主要设计思路及研究方向。并以工业化发展为前景,设计能够达到甚至超过工业基准的催化剂是电解水未来发展的焦点。随后,总结了构建大功率集成电极的方法以及催化电极的设计原则。最后,聚焦目前的工业化应用,就明析工业电解槽的设计瓶颈问题进行分析说明。
2.1 HER基元反应
碱性电解水主要涉及阳极析氧反应(4OH-→O2+ 2H2O + 4e-)和阴极析氢反应(4H2O + 4e-→2H2+ 4OH-)两个不同的半反应。与酸性环境相比,碱性条件下析氢需要经历缓慢的多步反应,其复杂的动力学变化导致HER活性评价指标复杂化,不再由单一的吸附氢(Had)决定20。实验结果表明,大多数电催化剂在碱性介质(pH = 13)中的HER活性比在酸性介质(pH = 1)中低2-3个数量级21,22。这个大的性能差异主要是由电解液中的H+浓度引起的。在酸性介质中,电解质中大量的H+被直接吸附在活性位点上。然而,在碱性介质中,需要一个额外的水解离过程来产生质子。碱性介质中H2O吸附比酸性介质中H3O+的吸附要弱得多,且涉及到强共价键H-OH断裂23,24。阴极HER是一种具有双电子转移的多步反应,步骤如下:
碱性环境,质子不参与反应,步骤如下25,26:
酸性环境,得益于电解液中的高质子环境,反应步骤如下:
此外,Tafel斜率也被普遍认同可以用来识别其可能的反应机理。例如,根据Butler-Volmer方程,塔菲尔斜率为38或116 mV·dec-1表明Heyrovsky或Volmer步骤分别作为HER速率限制步骤27。对Volmer-Tafel机理,由于Tafel过程是一个不涉及电子转移的化学反应,不遵循Butler-Volmer方程,因此Tafel斜率被认为是不变的(29 mV·dec-1)28。Tafel斜率越低,意味着相同电流密度的增加,所需的过电势越小,意味着反应动力学越快。Tafel斜率可以通过拟合Tafel图的线性区域来确定,这是得到Tafel斜率值最常用的方法。但是在大电流密度下,由于大量气泡的产生,电流密度通常会偏离线性关系29。
2.2 HER机制
与酸性条件下成熟的动力学活性描述符不同,普遍认为碱性条件下氢的吸附强度不再是绝对的评价标准,探索额外的水解离能垒以及碱性条件下催化剂对pH的依赖性不得不作为透析碱性HER机理的关键步骤之一。因此,尽管最近纳米技术的发展导致了许多高性能碱性电催化剂的产生,但其动力学缓慢的内在原因仍存在争议,碱性条件下HER活性位点的性质仍不确定。与酸性催化剂相比,碱性HER电催化剂设计原理的发展仍展现出巨大潜力。结合上述基元反应不难发现合理设计具有良好水和结合氢类解离能力的电催化剂能有效提高碱性HER性能30。根据Volmer和Heyrovsky步骤的反应途径,有四个主要因素可能影响碱性HER性能,即水吸附、水解离能力、氢结合能和OH的吸附强度31。近年来,被广泛认可的碱性HER机理主要包括氢结合能(Hydrogen Binding Energies,HBE)机制和双吸附位点机制(图1)。

图1 碱性HER反应机制示意图Fig. 1 Schematic diagram of different mechanism during alkaline HER reaction.
2.2.1 HBE质子理论
如图1a所示,HBE质子理论认为对于单金属活性位点,吸附氢(Had)作为唯一影响HER活性的描述符,被广泛认可30,32。催化剂表面的Had被认为是决定氢产生速率的重要因素。Had在优化后的表面既不太弱也不太强。如果Had太弱,在Volmer步骤中氢原子的吸附变得困难;
而Had强度过大,则在Tafel或Heyrovsky步骤中氢原子的解吸困难。为了更好地理解氢结合能的多功能性,Yan和同事33探索了一系列单金属表面上的HER活性与氢结合能之间的关系。如图2a所示,各种单金属催化剂的交换电流密度与H结合能形成了火山图,Pt作为公认拥有最优HER活性的金属,位于火山曲线的顶部,展现出最优的HER动力学。Cu/Ag/Au位于火山曲线的右分支,展现出弱氢结合能,氢结合能强的W/Fe/Ni/Co/Pd位于火山曲线的左边。最优的HBE可保持碱性介质中Had中间体的吸附和脱附平衡。最近对Pt在不同pH值下的HER活性的研究表明,电解质的pH值会通过改变催化剂的HBE来影响HER活性。Yan等34以多晶Pt为考察对象,研究电解质的pH值是否能够调控催化剂的HBE来影响HER活性。图2b为不同电解质下Pt(110)和Pt(100)晶面的CV曲线,随着pH不断提升(从0.2到12.8),欠电势沉积氢(Hupd)的峰位置展现出明显的pH依赖性。图2c则直接性地显示了pH的改变可以影响HBE,从而调控HER活性。随着酸性到碱性环境的改变,他们提出OH的吸附并不直接参与反应,而是通过OH浓度改变氢的结合能,进而决定了HER的整体性能。因此,他们认为,催化剂的HBE值是一种材料的固有特性,应该独立于局部环境中pH值的变化。为了进一步探索HBE机制的普适性,又进一步以负载型Pt族金属纳米颗粒(Pt、Ir、Pd、Rh)为研究对象35,研究表明各金属的HBE随pH呈线性增加,但斜率相似,说明pH值的变化对HBE的影响与金属无关。通过考察Pt/C、Ir/C、Pd/C、Rh/C的Epeak(分别在pH电解质范围为0-13的缓冲电解质中记录的CVs中获得,即HBE)与HER活性之间的相关性(图2d),发现了不同金属的Epeak与其表面的吸附强度EM-H之间普遍呈现出线性相关,从而进一步证明了HBE理论的普适性。

图2 (a)金属位点的HBE与交换电流密度log(i0)之间的火山图33;
(b)在不同pH电解质中,Pt(110)和Pt(100)晶面的稳态CV曲线34;
(c)Pt(110)(实心)和Pt(100)(空心)表面上的HBE作为溶液pH的函数34;
(d)负载型铂族催化剂的交换电流密度与Hupd峰位置之间的线性相关性35Fig. 2 (a)Volcano-type dependence between HBE and exchange current densities log(i0)on monometallic surface 33;(b)steady state CVs of Pt(110)and Pt(100)in different pH electrolytes 34; (c)HBE on Pt(110)(solid symbols)and Pt(100)(empty symbols)surfaces obtained from CVs as a function of solution Pt 34; (d)a correlation between the exchange current density and Hupd peak position for supported Pt-group nanoparticles 35.
然而HBE理论普遍认为H*是影响HER活性的单一因素,换句话说,认为除H*以外的中间体(如OH*和H2O*)只通过改变催化剂的HBE来影响反应。然而,Baek等36结合密度泛函理论(DFT)的计算发现Ru/C2N杂化物对OH的亲和力较低,将表现出更优异的碱性HER性能。Baek等认为如果OH吸附过强,活性位点将被最终产物占据,导致HER过电位升高。Goddard等37利用分子动力学模拟了不同条件下的碱性HER过程,得出结论,pH依赖的HER活性是由电极表面水吸附的变化引起的。在外加电场下,水的偶极会与电极之间产生相互作用,导致催化剂表面的吸附自由能(ΔGH2O)对pH具有依赖性。因此,对于HBE的影响机制仍是模糊不清的。而且HBE依赖的理论似乎并不是普遍的。例如,在Pt(111)表面,尽管HER活性在不同的pH下有很大的变化,但Hupd峰值的位置变化不大38。因此,许多研究人员认为,Hupd的性质不太可能单独由吸附氢来决定38-40。因此,考虑到碱性介质下催化剂表面复杂的界面结构关系,Koper等38提出了一个模型,在该模型中,Hupd的变化不仅是由于氢的吸附和解吸,还因为氢被O或OH取代。步骤中的O :OH比随步骤几何形状、步骤密度和介质而变化。与酸性介质中相比,碱性介质在该步骤中(或之上)吸附的OH相对较多。而在酸性介质中,在该条件下(或之上)吸附了更多的O,表明碱性HER的势垒主要取决于电极电位与零电荷电势(potential of zero free charge,pzfc)之间的关系。研究表明随着pH的不断变化,Hupd峰值的变化方式与pzfc相同。因此,将pzfc对氢吸附活化势垒的影响归因于界面水重组以适应双层电荷运动所导致的能量损失。在酸性环境中,Pt的pzfc接近氢吸附区,这意味着重组能量相对较小。而在碱性环境中,这样的能量与在酸性环境中相比要大得多。Koper等41通过在Pt(111)表面引入Ni氢氧化物沉积,pzfc可以位移到接近氢吸附区域的电位,Ni(OH)2通过降低界面水重组所需的能垒从而允许更有效的质子/氢氧化物通过双电层转移,从而提高了析氢速率。重要的是,该模型还暗示了氢结合能并不是HER动力学的唯一描述符,界面电场的强度也可以通过影响电荷转移过程中与界面水重组相关的能量势垒来影响氢的吸附速率31,39。这一结果从催化剂的角度也为下文提到的碱性HER电催化剂双吸附位点理论提供了新的理解思路。
2.2.2 双吸附位点理论
如图1b所示,与酸性环境下机制不同,在碱性环境下,往往认为第一步水解离成OH和H的快慢是HER的速控步骤,因此,碱性HER活性描述符除了H吸附能以外还涉及OH吸附能和水解离能42,43。然而与吸附Had相比,水的活化要缓慢得多。设计具有至少两种活性位点的催化剂将有效促进缓慢的碱性HER动力学,其中一种需要有利于水的解离,另一种具有中等的氢吸附自由能44。Markovic等45在金属Pt表面修饰了Ni(OH)2催化剂,研究表明Ni(OH)2的存在可以通过降低水解离的能垒来提高Pt的HER活性。Ni(OH)2团簇的边缘位点显示出优异的OH吸附能,加速水解离的同时还能与邻近的Pt活性位点表面吸附的H重组生成氢分子,从而实现快速的HER动力学。
为了进一步证实过渡金属氧化物的引入促进HER活性的双位点机制的普适性,Markovic等46将一系列常见过渡金属的氢氧化物(Ni、Co、Fe、Mn)修饰于金属Pt表面,进一步研究了HER活性与OHad-M2+δ键强度之间的相关性。通过一系列电化学测试发现OHad-M2+δ键强度与材料的HER活性呈负相关关系(图3a)。最近,McCrum和Koper41通过选择性地在Pt(553)单晶上沉积Mo、Re、Ru、Ag原子,构建了一种几乎恒定的Had结合强度和具有可变M-OHad结合强度的模型催化剂。他们发现,用DFT计算的OH结合强度,测定的碱性HER速率符合火山型曲线(图3b)。McCrum和Koper描绘了三维火山曲线(图3c),并证明了具有适度的Had和OHad吸附能的催化剂将表现出更好的催化剂性能。虽然关于HER中速率决定步骤(RDS)的动力学过程有很多争论,但目前流行的观点是涉及OHad的吸附或水解离的Volmer步骤是碱性HER中的RDS (图3d)。

图3 (a)Ni(OH)2/Pt(111)异质界面上碱性HER的示意图46;
(b)以Pt(553)为H吸附位点,研究的OH结合能与碱性HER活性之间的火山图;
(c)碱性产氢过程涉及H2O解离、OHad和Had吸附能的三维火山曲线;
(d)反应能图说明了HER的反应机理41Fig.3 (a)Schematic representation of the alkaline HER on a Ni(OH)2/Pt(111)heterointerface 46; (b)using Pt(553)as the H adsorption site, the volcanic diagram between OH binding energy and alkaline HER activity; (c)3D volcano curves considering dissociation energy of H2O and adsorption energy of OHad and Had during alkaline HER process;(d)reaction energy diagram to illustrate the HER reaction mechanism 41.
碱性HER双位点理论认为在碱性环境下,第一步水解离成OH和H的快慢往往是催化剂的速控步骤,因此,HER活性描述符还涉及OH吸附能和水解离能。因此,构建具有亲氧的活性组分的催化剂协同促进碱性HER动力学至关重要47,48。Ni-基材料作为活性亲氧组件,被广泛应用于与具有优异ΔGH吸附能的催化剂协同促进碱性HER催化进程。催化剂表面的氢氧化镍可以促进水的分解,并为H2的生成提供质子,使碱性HER的金属催化剂具有较高的活性。Kim等49通过加入一系列亲氧过渡金属原子(Ti、Cr、Fe、Ni),研究了镍基薄膜催化剂的HER活性随结合能的变化趋势(图4a)。研究发现,亲氧原子的掺杂可以调节镍表面H和OH的结合能力(图4b),从而首次建立了OH结合能与碱性HER活性之间的火山关系(图4c)。特别是,Cr掺入的Ni催化剂位于“火山”顶部,具有最优化的OH结合能和H结合能,以促进水的解离(RDS)和提高碱性介质中的HER活性。Gong等50发现Ni/NiO界面的构建导致了比纯Ni和NiO更好的HER活性。Hu等51也精心设计了一个Ni和NiO比例可调的Ni/NiO异质结构,其中NiO作为水的分解位点,Ni作为H2的生成位点,DFT计算表明,NiO表面的水解离能垒远低于Ni(111)表面的水解离能垒。Wang等51将单原子铂固定在NiO/Ni异质结构界面(PtSANiO/Ni)作为碱性析氢催化剂(图4d)。发现Pt单原子与NiO/Ni的异质结构使羟基(OH*)与氢(H*)的结合能力可调,有效调节水的离解能,促进H*的转化,加速碱性析氢反应。Li等52在CoS2上负载CeO2成功制备出纳米线阵列电极,其产生的界面路易斯酸碱Ce···S对具有独特的电子和结构配置,可有效激活水吸附解离并在动力学上加速碱性析氢,在10 mA·cm-2电流密度下供给了36 mV的低过电位。这种Ce···S对也削弱了CoS2上的O2吸附,赋予催化剂超过1000 h的未衰减活性的高稳定性。

图4 (a)掺杂亲氧金属(M)原子的Ni催化剂表面的碱性HER反应路径示意图;
(b)Ni(111)和M-Ni(111)表面的氢结合能(ΔEH)和羟基结合能(ΔEOH)的DFT计算对比图;
(c)ƞ10和Tafel斜率作为M-Ni和Ni催化剂ΔEOH的函数的火山图49;
(d)PtSA-NiO/Ni催化剂HER机理示意图 51Fig.4 (a)Schematic illustration showing reaction paths in alkaline HER on the surface of a Ni catalyst doped with oxophilic metal (M)atoms; (b)comparison plot of DFT calculation of hydrogen binding energy (ΔEH)and hydroxyl binding energy (ΔEOH)for Ni(111)and M-Ni(111)surfaces; (c)volcano plot of M-Ni and Ni catalysts with ƞ10 and Tafel slope as a function 49; (d)HER mechanism of PtSA-NiO/Ni 51.
然而,尽管基于双吸附位点理论的文章层出不穷,但是不得不考虑导致催化剂具有优异OHad吸附能的内在机制,以及双位点所引发的复杂的界面环境,以及外部电场等对催化剂的影响。
2.3 OER基元反应
与HER相比,OER涉及的反应途径更复杂、更缓慢,通常被认为是水电解过程中更需要提高热力学和动力学要求的反应。事实上,OER是羟基在碱性溶液中被氧化的过程。然而,由于OER的电子转移动力学过程复杂,因此尚未得出明确的反应机制53,54。目前,主要采用两种机制来解释OER过程:吸附氧演化机制(Adsorbate evolution mechanism,AEM)和晶格氧介导机制(Lattice oxygen-mediated mechanism,LOM)。AEM机制适用于解释各种催化剂在OER过程中活性位点的演变,而LOM更适用于各种高共价氧/氢氧化物55,56。
公式(7-10)是假设AEM涉及四个连续的质子-电子转移步骤,发生在单金属活性位点(M)上53,57:
如图5a所示,首先,一个羟基自由基(OH)被吸附在活性位点M上,通过一次电子氧化形成M-OH。然后,耦合的质子和电子从M-OH中去除,形成M-O*。第二个HOO-M生成步骤通常被认为是大多数催化剂的RDS。在这一步中,M-O*与另一个OH反应形成M-OOH。最后,M-OOH通过与OH发生脱质子作用,生产最终产物O2的同时使金属活性位点再生。

图5 (a)单金属位点的AEM途径示意图,黄色的代指金属位点,与金属相连绿色圆形表示晶格中的氧,绿色的球是电解质中的氧,黑色箭头代表加入,黄色箭头代表释放;
(b-d)氧空位、单金属、双金属分别作为活性中心的LOM机制,化学惰性晶格氧、涉及OER的活性晶格氧和电解质中的氧分别用绿色、红色和绿色球体标记,虚线代表氧空位Fig. 5 (a)AEM pathway on single metal site. Metal site, the lattice oxygen and oxygen from the electrolyte are marked in yellow, green color and green ball, respectively. The black arrow means join, and the yellow arrow means release.(b-d)LOM mechanism with oxygen vacancy, single metal and dual-metal as active site, respectively. Chemically inert lattice oxygen, active lattice oxygen involving OER, and oxygen in the electrolyte are marked with green, red, and green spheres, respectively, and dotted lines represent oxygen vacancies.
对于LOM途径,催化表面不再热力学稳定,会随着不断地氧演化过程发生动态变化58。因此,催化剂表面释放、交换、氧化晶格氧配体是提供OER循环的必要条件59。一旦晶格氧引入循环,OH/H2O物种将结合在氧空位位点,作为新的“晶格氧”进入下一个循环。因此,晶格氧激活是LOM途径能够成功触发的先决条件60,61。就LOM机制而言,其具体的OER路径是复杂多变的,就活性中心的不同主要分为以下几类60,62:①氧空位机制(图5b)63,64:活化晶格氧作为活性位点,通过亲核攻击直接接收OH形成*OOH物种,随后O2的释放产生了一个氧空位位点,并由OH重新填充,从而完成OER循环;
②单金属位点机制(图5c)65,66:以单个金属位点为催化中心来吸附OH,并遵循脱质子化步骤。表面重构使*O中间体与活化晶格氧直接耦合形成*OO物种,随后O2的释放同样会产生一个氧空位位点,并由OH重新填充,从而完成OER循环;
③双金属位点机制(图5d)67-69:相邻活化的晶格氧通过分子内氧偶联形成一个M-OO-M基序,其中形成的*OO*部分通常作为类似过氧化物的物种,随后释放出O2,形成两个氧空位位点,并由OH重新填充。
2.4 OER机制
电解水作为一种极具发展潜力的高效可持续产氢技术,缓慢的OER本征动力学严重阻碍了水电解的整体效率。因此深入了解OER的动力学机制,采取可行措施跨越OER动力学屏障至关重要。
传统AEM机制认为催化剂表面是静态的,活性位仅受到OH*和OOH*吸附能之间线性关系的限制(图6a)70,理论上最小过电位应为0.37 V。然而,许多已报道的先进电催化剂中OER的起始过电位比AEM提出的最小值要低得多61,71。并且,随着先进表征技术的发展,已有不少实验研究表明,催化剂表面在反应过程中会经历动态的结构演化,会导致表面重组的结构变化,特别是在高强度条件下72-74。因此,AEM机制下的OER动力学状态尽管可以通过引入第二个吸附位点、质子受体、阳离子、氢键等新变量促使OER催化剂动力学屏障跨越AEM机制下火山型曲线的顶部75,但是该机制并不适用于解释催化剂在OER过程中的动力学演变。近年来,晶格氧氧化还原衍生的LOM机制使具有高金属-氧共价性的固态电催化剂的高本征活性和表面重构问题合理化。在LOM机制下,晶格氧为媒介通过自身复合形成氧气分子,因此不经历OOH*中间物种生成,从而避免了线性关系的存在对催化剂活性提升的限制61,76,77。然而,LOM机制运行的前提条件是晶格氧的有效激活。如图6b所示62,68,78,图6b-①中TMd能带和O 2p波段中心之间的能量差呈正值,在热力学上,阳离子氧化还原电化学更有利于提供电子。电子转移发生在金属中心和吸附的氧中间体之间,遵循传统的AEM途径,而氧配体将被限制在晶格中而不被激活。图6b-②表明随着电荷转移能量的降低和U的增大,电子带可调,使LHB穿透O 2p带,将材料切换到负电荷转移绝缘体处。因此,在晶格基体中,从氧配体到TM阳离子的分子内电子转移是可行的,留下配体空穴用于晶格氧激活63,79,80。这种电子调制也可以改变OER中间体的化学亲和性,从而降低能量势垒,即使晶格氧不能直接氧化为氧分子,也可以通过促进水亲核攻击来优化AEM机制动力学,从而提高OER活性81-83。如图6b-③所示,为了使晶格氧参与OER过程实行LOM机制,O 2p带需要上移至费米能级附近,促使其与金属的d带中心轨道重叠(即增强M-O键的共价性),赋予晶格氧的氧化还原足够的能量跨越。与O2/H2O的氧化还原相比,还需要调节O 2p带的绝对能级,越过费米能级,具有大量的空穴才会使晶格氧不稳定,从而实现氧的释放60,79,84。

图6 (a)氧上水解离的自由能(ΔG*OOH - ΔG*O (eV))与质子去除自由能(ΔG*O - ΔG*OH (eV))的比例关系 70;
(b)在 d-d 库仑相互作用(U)和电荷转移能(Δ)的引导下的阳离子/阴离子氧化还原化学示意图,表现为传统的金属阳离子氧化(左)、阴离子氧化(中间)和直接氧阴离子释放(右);
(c)NaxMn3O7在|O 2p孤对态中的氧空穴形成示意图;
(d)NaxMn3O7的投影态密度(x = 2、1.5、1、0.5)79;
(e)以Mott-Hubbard为基准,计算的CoO2和锌取代CoO2板的能带示意图;
(f)Na+引入导致的OER机制的转变,包括AEM (左)和LOM (右)84Fig. 6 (a)The scaling relationship between the free energy of the water dissociation on top of oxygen (ΔG*OOH - ΔG*O (eV))and the free energy of proton removal (ΔG*O - ΔG*OH (eV))70; (b)The schematic representations of cation/anion redox chemistry guided by d-d Coulomb interaction (U)and charge transfer energy (Δ), which manifest conventional metal cation oxidation (left), oxygen anion oxidation (middle)and direct oxygen anion release (right)for OER, respectively; (c)Schematic formation of oxygen holes in |O 2p lone-pair states for NaxMn3O7; (d)Projected density of states of NaxMn3O7 slabs (x = 2,1.5, 1, 0.5)79; (e)Schematic energy bands of CoO2 and zinc-substituted CoO2 slabs in consideration of Mott-Hubbard splitting; (f)Transformation of OER mechanism due to Na+ introduction, including AEM (left)and LOM (right)84.
最近,Wang等79提出通过碱金属离子(Na+)介导调节晶格氧反应性和标度关系来构建更好的氧演化电催化剂的有效策略。具体来说,通过调节Mn空位形成的氧孤对态中的氧空穴的数量来决定Na+的数量与晶格氧反应性,同时可以控制O-H键裂解和O-O键形成之间的势垒对称性。另一方面,Na+的存在可以与*OOH中的垂体氧发生特异性的非共价相互作用,以克服线性标度关系的限制,降低过电位。如图6c所示,首先利用密度泛函理论(DFT)计算并解释了激活晶格氧的契机是自旋特征的配体空穴。根据电荷转移能(Δ)和dd库仑相互作用(U)的振幅,Na2Mn3O7(U > Δ)位于电荷转移状态,在完全填充的|O 2p带上方显示一个空的金属带。并且随着Na+数量的减少,激活晶格氧时,|O 2p产生更多的氧空位(图6d)。简单来说,将Na2Mn3O7催化剂中的Na+移除,能够有效激活晶格氧释放O2,从而将OER运行机制从AEM转为LOM机制,不仅打破了AEM的*OOH和*OH中间体对垒所带来的高过电势屏障,而且通过优化氧空位的数量,能够有效促进晶格氧机制的进行。同样地,Wang等84通过在CoOOH中引入不同含量的催化惰性Zn2+,对LOM机制的形成条件进行了深入研究。Zn2+的引入导致了不同局域构型的氧非键态,构造了更多的氧空穴(图6e),随着Zn含量的不断提升,Zn-CoOOH的OER催化机制逐渐从AEM转变为LOM机制(图6f)。目前大量的方法已经被实施来微妙地调节电子态,以达到触发TM氧化物和(氧)氢氧化物中的晶格氧氧化还原化学的目标,包括构建阳离子缺陷工程,高价金属掺杂,晶格收缩,自旋翻转,表面重构,无定形等等53,61,85-88。但是,LOM机制也表明了电催化剂本身的亚稳态表面,允许不可逆的结构重建。由于电化学反应发生在催化剂/电解质界面,是重构表面提供了真正的催化中心。因此,针对性的构建界面模型,模拟界面化学在OER过程中的动态变化才能更真实的捕捉催化过程真正的活性位,从而进一步提升催化剂的稳定性。而对于适用于大部分催化剂的AEM机制,进一步探索打破*OH与*OOH竞争线性关系的策略是有效跨越动力学壁垒,降低过电势的有效手段。此外,Sun等81报道了一种微孔聚合物(Aza-CMP)配位单钴位点(Aza-CMPCo)OER催化剂,其呈现出一种分子内羟基亲核攻击(IHNA)路径,相对于常见的酸碱/水亲核攻击(WNA),这一过程导致活化能降低,动力学加速。
开发高效、稳定的双功能水裂解电催化剂具有重要意义,但具有挑战性89,90。深入了解电解水动力学障碍,能够有效地从原子水平构效关系上指导发展先进的电化学催化剂。基于上述机制,越来越多的研究人员开始聚焦活性位点的构建,以及它们如何有利于水分子或中间体的优化吸附和解吸(*H、*O、*OH和*OOH)。近年来,过渡金属化合物被广泛用作碱性水分裂的高效双功能电催化剂,如金属氧化物/氢氧化物91,92、磷化物93、硫化物94、硒化物95-97,等等。缺陷/空位工程98、异质界面48,96,99、杂原子参杂100,101等已被广泛地用于引入催化剂中的活性相从而提高催化剂本征活性。另一方面通过探索表面形貌工程、尺寸效应等来提高活性位点的密度也是有效提高催化剂电解水活性的有效手段102,103。
Zhang等104采用直接热解法在氮掺杂碳纳米管中封装Co2P-CoN双活性位点(Co2P/CoNNCNT),构建异质结构核壳纳米颗粒(图7a)。图7b中可以明显观察到分别属于Co2P和CoN的晶格条纹,以及碳纳米管的高结晶度。进一步DFT计算表明吡咯N被认为与HER活性有关,因为只有一对孤对电子作为活性催化中心(图7c)。得益于异质结构带来的双活性位,Co2P/CoN-NCNT对HER和OER均展现出优异的电化学活性。密度泛函理论计算和实验结果都表明,吡咯氮与Co2P的耦合展现出HER的高效催化,而CoN和氮掺杂碳壳界面上的Co-N-C活性位点负责该催化剂的OER活性。杂原子B作为电子供体,它的存在可以对金属原子产生一定的亲电作用力,从而提供额外的结合位点,如金属-硼-氢键。Zhang等105利用通过自组装硼酸链的简单原位热解工艺(图7d),合成了分散在B、N掺杂碳纳米纤维上的新型二三硼钌纳米颗粒(Ru2B3@BNC)。图7e为Ru2B3@BNC在碱性环境下的电化学HER性能,在10 mA·cm-2电流密度下仅需为7 mV的超低过电位,已经优于常用商业Pt/C以及目前报道的大多数HER催化剂。结合DFT (图7f)的计算结果,揭示了缺电子B原子可以促进局部电子再分配,从而导致了H*对Ru原子对Ru2B3中Ru-B键的锚定偏好效应。Mu等106通过自上而下的合成策略实现了分层分支Mo掺杂硫化物/磷化异质结构(Mo-Ni3S2/NixPy空心纳米棒)的合成。其优化的电子结构、分层支状空心纳米棒结构和丰富的非均匀界面赋予了多位点Mo-Ni3S2/NixPy/NF电极在碱性环境下展现出显著的稳定性和双功能电催化活性。将Mo-Ni3S2/NixPy同时作为阳极和阴极组装成碱性电解槽,它只需要1.46 V的超低电池电压,即可达到10 mA·cm-2的电流密度,具有优异的耐久性超过72 h,性能优于大多数报道的镍基双官能团材料。密度泛函理论结果进一步证实了金属掺杂异质结构可以协同优化HER过程中H*和含O中间体(OH*、O*和OOH*)的吉布斯自由能和OER过程,从而加速电化学水裂解的催化动力学。
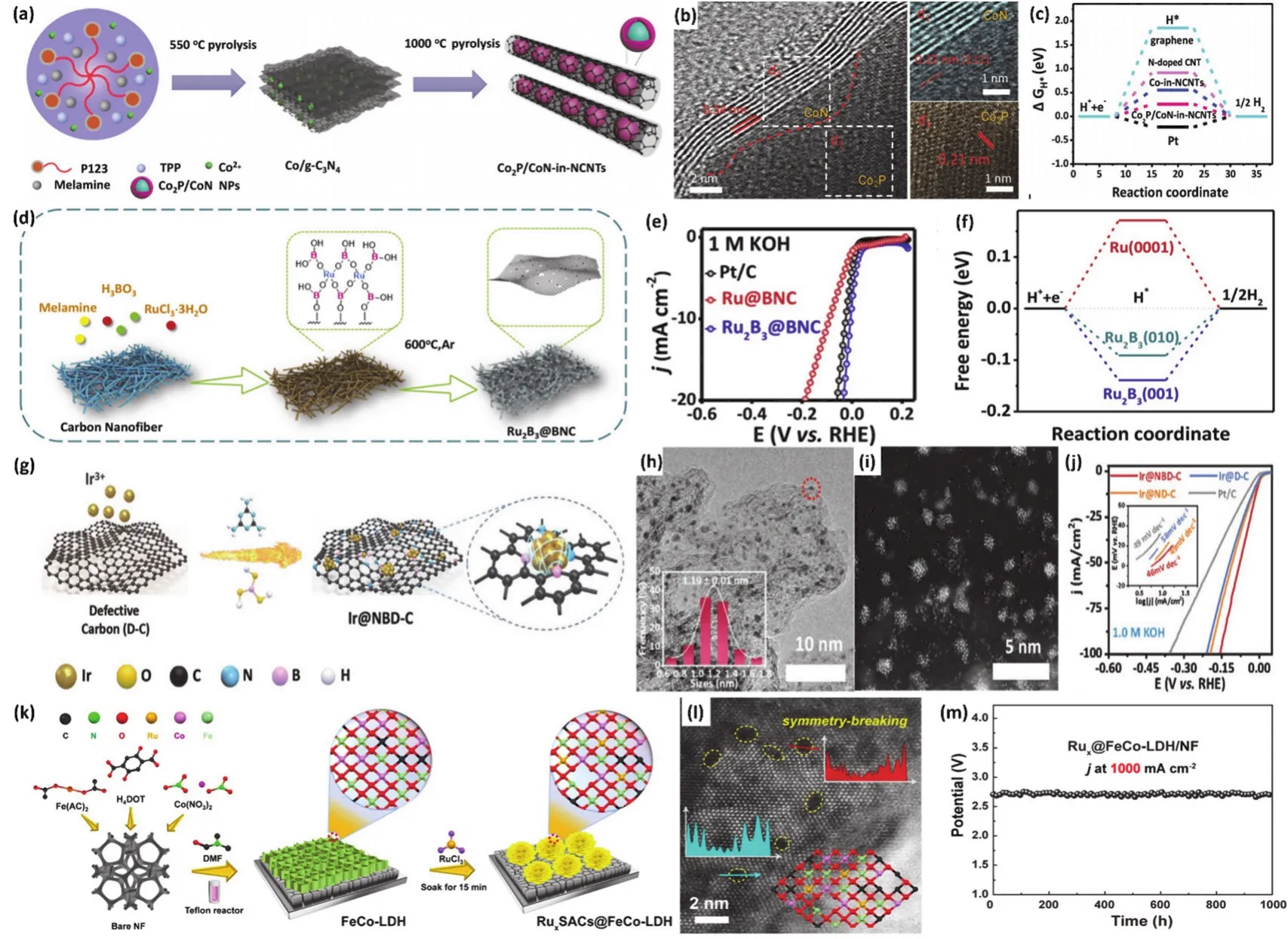
图7 (a)两步热解法制备Co2P/CoN-NCNT的合成路径示意图;
(b)Co2P/CoN-NCNT核壳结构的HRTEM图;
(c)不同催化剂的自由能图104;
(d)Ru2B3@BNC的合成步骤示意图;
(e)Ru2B3@BNC催化剂的HER活性;
(f)Ru2B3模型的氢吸附自由能105; (g)Ir@NBD-C催化剂的合成示意图;
(h-i)Ir@NBD-C催化剂的TEM图和HAADF-TEM图;
(j)Ir@NBD-C催化剂及对比样的LSV曲线112;
(k)Ru1SACs@FeCo-LDH催化剂的合成及微观结构设计示意图;
(l)FE-SEM图像,黄色虚线圈出了缺陷强度曲线;
(m)全解水过程RuxSACs@FeCo-LDH电极的长期稳定性119Fig. 7 (a)Schematic of the synthesis route of Co2P/CoN-NCNT prepared with two-step pyrolysis process; (b)HRTEM images of Co2P/CoN-NCNT core-shell structure; (c)Calculated free-energy diagram of different catalyst samples 104;(d)Synthesis process of Ru2B3@BNC catalysts; (e)HER activity of Ru2B3@BNC in 1 mol·L-1 KOH; (f)Calculated free-energy diagram of H adsorption 105; (g)Synthesis process, (h)TEM image and (i)HRTEM image of Ir-NBD-C;(j)LSV curves and Tafel slopes of Ir-NBD-C and other samples 112; (k)Synthesis and microstructure design of Ru1SACs@FeCo-LDH catalyst in alkaline; (l)FE-SEM image with the yellow dashed line showing the defect intensity curve; (m)Long-term stability measure of RuxSACs@FeCo-LDH catalyst during overall water splitting 119.
大量的实验研究表明,将金属催化剂的尺寸调整到纳米尺度可以显著地促进可接近活性位点的数量。然而,这些粒径较小的催化剂颗粒往往具有较高的表面能,纳米颗粒严重聚集,导致电催化活性显著下降22,107,108。因此,不仅需要进一步探索小尺寸金属颗粒的创新,还需要开发具有稳定锚定金属活性相的支撑材料。近年来,负载型高分散催化剂因其最大的原子利用率而被广泛应用于储能、绿色催化等众多反应中109,110。大量研究表明,高分散活性表面对于提升催化剂表观活性至关重要,然而,高分散表面的高表面能会促使金属团聚,从而大量损失活性位,致使金属大面积失活108,111。Zhang等112探讨了电子供体硼(B)束缚工程与铱(Ir)之间的共轭效应,促进了Ir原子的电子捕获,实现了高度分散的Ir局限于N、B共掺杂缺陷碳(图7g)。图7h,i可以观察到均匀分散于碳基质中的Ir纳米颗粒,Ir的平均直径仅为1.19 ± 0.01 nm。得益于均匀分散的小尺寸纳米颗粒赋予的大量可及活性位点,以及束缚工程对于颗粒的锚定作用,Ir-NBDC展现出优异的碱性析氢活性(图7j,ƞ10= -7 mV)。
2011年,Zhang等113首次制备了单原子催化剂,单原子催化剂的概念为电催化的进一步发展提供了方向,为设计和构建高效电化学催化剂提供了新的平台。根据中心金属原子的数量,原子分散催化剂可以分为几种不同的类型,包括单原子催化剂(SACs),双原子催化剂(DACs)和团簇。近年来,随着先进表征手段的发展,在明析催化剂表观原位动力学的基础上,单原子催化剂的合理设计与制备为显著提高水裂解的电催化活性提供了一种很有前途的策略。考虑到碱性环境HER/OER复杂的机制,单一的位点很难高效催化HER/OER动力学。因此,相较于单一单原子位点,双金属位点、以及和团簇以及颗粒协同作用更有利于HER/OER动力学114-116。近年来,越来越多的负载型单原子催化剂选择已有优异催化活性的Ni、Fe、Co基催化剂作为载体,在此基础上对其进行改性,调整其电子结构的同时引入了更多的催化位点,成功运用于碱性电化学产氢117-120。其中,Mu等119采用一种简单方法将亲氧金属钌(Ru)单原子稳定化构筑在对称性破缺的FeCo-LDH电催化剂(RuxSACs@FeCo-LDH)表面(图7k),该催化剂的构建为解决碱性条件下大电流全解水制氢的低效率和不稳定性提供了新的设计思路。FeCo作为电解水催化剂常用金属材料,其复杂的金属衍生复合物构建的丰富表面缺陷位点可有效锚定单原子催化剂,利用单原子催化剂的高原子利用率和FeCobased催化剂的高活性,整体促进催化剂的活性位点数量和电荷转移速率。但是距离达到工业级电解水的应用要求,仍然还有很大的距离(电流密度≥ 500 mA·cm-2,电压< 1.8 V)。RuxSACs@FeCo-LDH催化剂利用Ru单原子部分取代金属Fe(Co)在打破FeCo-LDH结构对称性的同时引入大量缺陷位(图7l)。该策略通过引入单原子对催化剂的金属-金属界面相互作用和电子环境进行调整,不仅促进了原子-界面-电解质之间的电荷转移,还极大地暴露了活性位点,使得该催化剂展现了及其优异地OER/HER活性及大电流密度下全解水性能。在碱性条件下,该催化剂驱动1000 mA·cm-2电流密度OER和HER的过电位仅分别为246 mV和117 mV,并且该电位下持续稳定1000 h无明显衰减。将其组建成碱性全解水电解槽,在1.52 V的超低电压下即可达到1000 mA·cm-2的工业级电流密度,同样经1000 h测试后,电压衰减几乎不计(图7m)。此外,具有高活性位点密度的非晶化表面,引入缺陷,如氧空位,相界,原子畸变,应变,孪晶,晶界/位错,堆积缺陷,和暴露低配位表面缺陷等所导致的电子结构优化和对反应中间体结合强度的影响探索也已成为一种很有前途的电催化剂设计策略108,121-125。
然而,大多数催化剂通常是在较低电流密度下进行(< 200 mA·cm-2),为了加快工业化应用脚步,探索电流密度大于500 mA·cm-2下稳定高效的电解水催化剂至关重要4。通常地,在高电流密度下,电极上总能产生大量的H2/O2气泡。电催化剂与电解质界面上气泡的积累会严重阻碍液体的传质,减缓电子的转移,降低来活性位点的暴露密度,导致电催化活性和耐久性变差4,90,126,在此条件下,低电流密度下稳定高效的催化剂并不完全适用于工业型应用。因此,洞悉设计有效的大功率电催化剂的瓶颈,针对设计高效稳定催化剂应用于电解槽是下一步需要集中解决的实际问题。
4.1 三维电极
电极的制备通常包括用聚合物粘合剂将催化剂粉末粘接或喷涂到集流器上的过程。粉末催化剂的增材制造工艺需要额外的设备和复杂的工艺,从而增加了成本。同时,这一过程增加了电荷转移阻力,导致催化剂在高电流密度下容易基质分层,限制了电解水装置的性能18,127。在高导电性衬底(Ni foam、carbon、Ni foam、Cu foam等)上原位生长的电催化剂纳米阵列将为克服上述限制提供另一种解决方案,可以提高实际催化性能。三维电极材料直接作为催化剂进行碱性电解水产氢展现出极大的优势28,93:1)避免可能有害的粘合剂的涉及;
2)操作简单,能够快速直接制备工作电极;
3)良好的机械黏附确保了活性相与导电基底的紧密接触,与电解液的直接接触,提高材料整体导电性的同时,加速了电子传递速率;
4)暴露更多的比表面积,增加活性位点数量;
5)针对大电流密度,三维材料的高传质通道能进一步加快催化剂表面气泡的释放,加快传质。虽然三维电极的设计已经取得了很大的进展,但仍然存在挑战。首先,受导电载体本身结构和物理化学性质影响,活性相的原位生长速率和形貌以至于最后呈现出来的电化学活性都不尽相同,测量的三维电极的表观活度实际上来自于活性材料和衬底。因此,很难对活性相真实催化位点活性进行精准评估。活性相的负载量对催化剂整体电化学活性也有很大的影响,尤其三维泡沫基底其内在的多孔疏松的网络结构,很难精确定量活性材料的负载。并且,金属相导电基底在电化学测试过程中与活性相交织的表面重构也是复杂多变的128,129。此外,现有的方法通常只生产面积很小的电极(< 10 cm2),比商业电极的面积小10倍。
4.2 规模化合成
因此,尽管碱性全解水催化剂的设计和开发取得了长足的进步,但仍存在一些阻碍其商业应用的巨大障碍,比如缺乏大规模合成催化剂的方法;
活性物质易从基底剥离;
催化剂表面气泡动力学;
在高电流密度下稳定性差等。催化剂的进一步发展需要低成本、绿色、易操作、重复性高和可持续的合成化学。目前碱性电解水产氢的规模化合成的研究却很少。大量报道的合成方法还局限于实验室水平,限制了这些催化剂的实际应用。对于工业PEM和碱性电解槽,电极尺寸一般为100 m2,而在实验室中使用的电极尺寸一般为10 cm2或更小。小电极尺寸导致电场分布不均匀,大电极的质量输运行为不同,因此小电极上的催化性能不能转化为大电极尺寸上的催化性能还有待商榷。诸如电场分布、质量传输效率和热管理等问题,应该考虑用于大电流密度下水分离的面积电极。此外,还需要更高效省时的稳定性和耐久性试验方法。
Pint等130利用纳米材料的二元溶液和液体电泳组装,快速构建电池设计应用的混合材料。如图8a-c所示,他们利用电泳沉积原理开发了一种全自动的台式滚动系统。从溶液中获得的滚动电泳组装使这些纳米结构的均匀涂层得以可控地制造,以保持纳米材料本身的化学和物理性质,并且也没有使用表面活性剂或杂质的不利影响。但是,目前该种方法并未运用于电解槽工业电极的制备,是值得参考的一种低成本、快速、有效的大规模合成技术。Liu等5成功地合成了廉价、高效和耐用的MoS2基电催化剂,并且其在工业要求的高电流密度下(1000 mA·cm-2)均能够工作良好。如图8d所示,MoS2基电催化剂采用两步法合成,首先通过MoC2作为中介,利用中间辅助磨削剥离(iMAGE)技术剥离大块二硫化钼,然后将2D MoS2薄片粉末分散在水中,获得催化剂分散墨液。这种分散均匀的墨状催化溶液适合运用于各种成熟的工业使用技术(电气浸涂、滴铸、滚辊印刷、丝网印刷、喷涂等),以生产大面积集成电极。以0.5 m2Cu泡沫为基底,成功合成了MoS2基电催化剂,该催化剂在高电流密度下仍然表现出优异的电化学活性,1000 mA·cm-2的高电流密度仅需要412 mV的过电位(图8e)。随后,得益于三维材料电极的优势,Liu等131进一步以Ni泡沫为基质,由氢氧化物介导的Ni4Mo纳米颗粒修饰FeOx,并固定在MoO2纳米片上合成了该催化剂(h-NiMoFe)(图8f),用于高电流密度析氢,并且仅需97 mV的超低过电位即能在1000 mA·cm-2的电流密度下运行(图8g)。Zhai等126提出了一种由分散的NiFe氢氧化物纳米粒子和超薄NiS纳米片(NiFe LDH/NiS)组成的肖特基异质结纳米片阵列,以协同调节OER高电流密度下的质量输运和电子结构。富孔隙度的NiS纳米片阵列为快速传质提供了丰富的催化位点和良好的电解质渗透。NiFe LDH与NiS的耦合可调谐的d波段中心Ni(Fe)原子与氧中间体的结合强度为有利OER动力学。NiFe LDH/NiS肖特基异质结表现出了显著的OER活性,在1000 mA·cm-2的电流密度下仅需要325 mV的超低过电位。

图8 (a-c)利用电泳沉积原位设计催化剂的滚动装置及原理示意图130;
(d)MoS2基油墨型电催化剂的高通量生产合成步骤及结构示意图;
(e)MoS2基油墨型电催化剂的LSV曲线5;
(f)h-NiMoFe催化剂两步法的合成过程与形貌演变;
(g)h-NiMoFe催化剂的HER活性126Fig. 8 (a-c)Schematic of the roll-to-roll system and design principle of catalyst prepared by electrophoretic deposition 130; (d)diagram of high-throughput production step and structure of MoS2-based ink-type electrocatalysts; (e)LSV curves of MoS2-based electrocatalysts 5; (f)synthesis and characterization of the h-NiMoFe catalyst; (g)LSV curves of h-NiMoFe, Ni foam and commercial Pt/C 126.
4.3 亲水疏气特性
理解三相界面(即固体催化剂、液体电解质和气体产物)对高性能催化剂的设计至关重要。例如,工业电解槽通常在非常大的电流密度下运行(> 200 mA·cm-2)。如图9a所示,在大电流密度下,催化剂表面会快速产生大量气泡。气泡的产生会导致一系列催化问题4,132,133:1)气泡附着于催化剂表面会阻碍了催化剂与电解质界面的传质速率;
2)气泡层的厚度随着电流密度的增加而增加,附着在催化剂表面的气泡会覆盖掉大部分催化剂表面,降低了其催化性能;
3)由于附着在催化剂界面上的气泡在离开催化剂时会对催化剂产生很强的界面附着力,导致部分催化剂可能会被气泡剥离,从而降低催化剂的机械稳定性。除非催化剂与载体之间的相互作用力大于催化剂与气泡之间的界面附着力,否则随着电流密度的增加,这种催化剂剥落问题通常会变得严重。因此采用三维无粘连剂催化电极能极大程度上稳定催化剂。此外,构建超疏气电极可以降低气泡粘结力,加速气体的释放,有利于增强质量输送,从而提高电极性能。
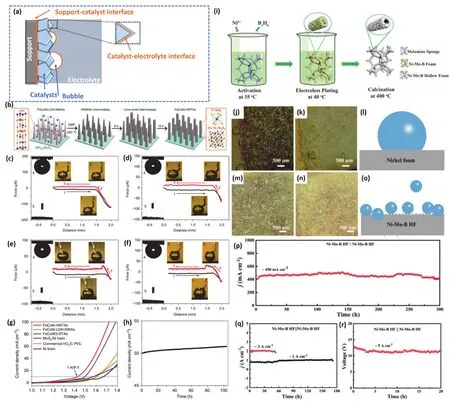
图9 (a)催化剂表面产生气泡示意图;
(b)FeCoNi-HNTAs催化剂的合成步骤及形貌变化示意图;
(c)FeCoNi-HNTAs,(d)FeCoNi-LDH-HWAs,(e)MoS2/NF和(f)NF在电解质中的超疏气和超亲水性测量;
(g)催化剂全解水性能对比;
(h)FeCoNi-HNTAs的全解水稳定性134;
(i)NiMoB-HF/NF中空纳米管状催化剂的合成示意图;
(j)低电流密度(20 mA·cm-2)下和(k)大电流密度(100 mA·cm-2)下的H2气泡附着在NF上的光学照片;
(l)气泡附着在NF表面的示意图;
(m)低电流密度(20 mA·cm-2)下和(n)大电流密度(100 mA·cm-2)下的H2气泡附着在NiMoB-HF/NF上的光学照片;
(o)气泡附着在NiMoB-HF/NF表面的示意图;
(p-r)不同电流密度下NiMoB-HF/NF催化剂的长期稳定性135Fig. 9 (a)Diagram of the formation of bubbles on the catalyst surface; (b)Schematic diagram of synthesis steps and corresponding morphologies of FeCoNi-HNTAs catalyst; Superaerophobic and superhydrophilic measurements of(c)FeCoNi-HNTAs, (d)FeCoNi-LDH-HWAs, (e)MoS2/NF and (f)NF; (g)Comparison of overall water splitting performance of catalysts; (h)Long-term stability measurement of FeCoNi-HNTAs 134; (i)Illustration of synthesis of NiMoB-HF/NF hollow nanotube catalyst; Optical photos of H2 bubbles attached to Nickel foam (j-k)and NiMoB-HF/NF(m-n)at (j, m)low current density (20 mA·cm-2)and (k, n)large current density (100 mA·cm-2), respectivity;Schematic diagram of bubbles attached to Ni foam surface (l)and NiMoB-HF/NF (o). (p-r)Long-term stability measurement of NiMoB-HF/NF at different current densities 135.
与传统的滴铸法制备的平面电极相比,超疏气电极对气泡的低附着力,加速了气泡的演化,导致气体演化反应电流快速、稳定。Sun等134设计合成了由过渡金属硫化物(FeCoNi-HNTAs)组成的用于整体水分裂的超疏气纳米管阵列电极(图9b)。FeCoNi-HNTAs在电催化HER (电流密度为10 mA·cm-2下仅需58 mV的过电位)和OER (电流密度为10 mA·cm-2下仅需184 mV的过电位)中均展现出优越的电化学活性和长期耐久性(200 mA·cm-2电流密度下能连续稳定80 h以上)。为了便于确定纳米阵列从电极表面释放气泡的结构优势,进一步研究了制备电极的表面超疏气性。如图9c-f,对不同结构催化剂进行了电解质环境下的超疏气和超亲水性测量。FeCoNi-HNTAs、FeCoNi LDHNWAs、MoS2/Ni和Ni泡沫的气泡粘附力测量。图中插图1-3显示了相应的粘附力测量过程中的气泡状态,其中过程1表示电极表面接近气泡,过程2显示电极表面离开气泡,工艺3显示与气泡分离的电极表面。在图9c和d的插图2中能观察到的明显的气泡变形,进一步证明了FeCoNi-HNTAs和FeCoNi LDH-NWAs明显没有气泡粘附力。而在MoS2/Ni泡沫(10.6 ± 1.91.9 μN)和Ni泡沫(16.0 ± 1.7 μN)上测量出较大的气泡粘附力,主要得益于Fe、Co和Ni离子之间的协同效应,以及丰富的催化活性位点和1T’MoS2的超导电性。另一方面,电解质下的超疏气性也有利于形成的气泡从电极表面脱离,有助于显著提高全解水活性及稳定性。图9g-h显示了FeCoNi-HNTAs催化剂优异的全解水活性(10 mA·cm-2的电流密度中的电池电压仅为1.429 V)及稳定性(50 mA·cm-2电流密度下能连续稳定100 h以上)。Shen等135以NiMoB/NF催化剂为媒介进一步研究了催化剂表面的气泡动力学(图9i)。图9j-l分别展现了低电流密度(20 mA·cm-2)和大电流密度(100 mA·cm-2)下的H2气泡附着在镍泡沫上的光学照片及说明。在NF电极的HER测试中,可以观察到一个相对较大的直径为≈ 500 µm的H2气泡。图9m-o可以发现Ni-Mo-B/HF电极释放出一个50-80 µm的H2气泡,小气泡有利于更快地H2解吸速度。小气泡引起的小拉伸力会使电极的损伤最小化,从而延长电极的使用寿命。全解水稳定性测试数据(图9p-r)进一步证明了Ni-Mo-B/HF电极的超疏气性有利于最小化催化电极损失。
因此,采用大规模工业合成技术制备超疏气三维电极能够有效促进催化剂表观动力学,以及大电流密度下界面气泡动力学所导致的缓慢传质速率活性相脱落以及催化电极活性和寿命下降等问题。
4.4 工业催化电极设计进展
质子交换膜电解槽(PEMWE)、碱性阴离子交换膜电解池(AEMWE)以及常规碱性水电解(AWE)是三种低温(< 100 °C)水电解槽。其中,AEMWE技术相对最不成熟,不仅是由于其发展较晚,投入产业化仍需时间,其次是由于其在投入实际应用前仍需进一步对AEMWE催化电极进行设计。因此,将目前探索成熟且具有高活性及高稳定性的镍基催化剂进行实用型设计运用于AEMWE,对于碱性电解槽的快速发展至关重要。然而,要想进一步将发展成熟的大电流密度催化剂成功运行工业电解槽,需要对电解槽核心组件—催化电极进行设计。传统的AEMWE电极由两部分组成:催化剂层和GDL层。除了考虑实际工作电极,扩散层电极材料对于整体电解水工作效率及寿命也至关重要。在实际电堆中,阳极高腐蚀性迫使GDL层材料多为金属基材,其中以Ti板实用性最高。而阴极环境的腐蚀性较小,一般使用成本效益高的碳基材料(例如,碳纤维,碳纸,或碳布)作为GDL层136。因此,考虑到大电流工作环境对催化剂及槽压及欧姆损耗的影响,针对AEMWE的HER/OER电催化剂的制备往往集中在粉末催化剂上。然而,催化剂直接沉积或生长于多孔和高表面积电流集成器137,用于制备统一性工作组件设计方法也受到了关注,这种设计可以使AEMWE操作进入高电流密度(> 5 A·cm-2)状态。
统一性工作电极可以适当地将催化剂与GDL层均匀复合在一起,加速气泡扩散的同时也防止催化剂粉末从GDL层脱落。Sung等137提出了一种三维均一电极,由镍铁氧氢氧化合物(NiFeOOH)直接电沉积在气体扩散层上构成,开发作为AEMWE阳极催化剂。图10a对传统电极和统一性电极材料进行了简单介绍。传统的电极材料多为催化剂颗粒(NP)喷涂于GDL层组成的密集电极,其二级孔约为10-200 nm。图10b-c对比介绍了使用统一电极和使用IrO2NP制备的常规AEMWE的性能。NiFeOOH在1.9 V电压下表现出优异的电流密度性能3600 mA·cm-2,并且最高电流密度可以达到5 A·cm-2。此外,尽管使用了非贵金属催化剂,但统一电极AEMWE的性能比传统的贵金属催化剂高2倍以上,同时具有更低的电荷转移和欧姆电阻。进一步说明电极设计赋予AEMWE电解槽高活性及工业可及性的同时提高了其高经济效益,加速产业化。由于商用AEMWE复杂运行过程离子通过膜的传导引起的欧姆损失以及由阳极和阴极过电位引起的活化损失导致AEMWE催化剂出现明显的电压损失,稳定持续下降138。因此,到目前为止,许多研究的发展重点要么是提高AEM的电导率,要么是研究高活性催化剂。AEMWE通常表现出较低的耐久性。NiFeOOH在工业电流密度下(500 mA·cm-2)持续稳定了100 h (图10d)。在此之前也有不少统一性电极以及喷涂型催化剂被设计合成,表1中对此进行了归类总结。

图10 (a)统一性与传统AEMWE电极两种类型电极的示意图;
(b-d)两种类型电极的AEMWE性能137Fig. 10 (a)Schematic diagram of two types of electrodes(Conventional and unified electrode with the Ti paper);(b-d)AEMWE performance of LSV curves,Nyquist plot and stability 137.

表1 AEMWE电极性能介绍Table1 Introduce of electrode performance of AEMWE.
5.1 常用电解槽介绍
为了对当前电解槽性能及产业化程度进行初步了解,我们首先对目前发展的四种不同的电解槽进行简单介绍,如表2所示145,146。在应对市场化方面,AWE作为发展最早、最为成熟的电解水产氢技术占据着主导地位,尤其是一些大型工业项目的应用。但是其效率低且危险性大,AWE采用石棉作为隔膜,其隔膜多孔材料有氢气渗透风险,导致大密度氢氧混合。并且,AWE的电解质主要为氢氧化钾,其在大气压下的120 °C下开始沸腾。因此,AWE倘若在高温下运行,温度超过120 °C,必须在高压下进行。报道显示高压状态下进行高温运行能够降低电池整体运行功率,但对电加热器、电动热泵(EHP)系统等装置平衡(BOP)的需求功率也相应增加147。

表2 常用电解槽特性介绍Table 2 Introduction of common electrolytic cell characteristics.
质子交换膜电解槽(PEMWE)中关键组件PEM膜传导质子,能够有效阻隔电极两侧产生的气体,从而避免AWE使用强碱性电解质以及多孔隔膜所伴生的缺点。PEMWE电解槽一般采用零间距结构,其不仅能够有效降低整个电解槽的欧姆电阻,且在高电流密度下能有效避免传统电解槽由于气泡造成的板间直接表面积的损失;
压力调控范围大(氢气输出压力可达数兆帕),并且可以快速与间歇性能源产生响应(< 5 s)。但是受限于PEMWE的酸性腐蚀环境、阳极高电位、传质速率等要求,PEMWE所使用的的电催化剂材料主要是以Ir、Ru等贵金属/氧化物及其合金/混合氧化物为主体,钛材料为载体的负载型催化剂,因此成本相对较高。
新一代AEMWE不仅可以有效解决传统电解池的缺陷,而且能够灵活地使用低成本、高储量非贵金属电催化剂制备膜电极作为隔板,单从操作特征看其展现出优异的发展潜力,但是目前还未产业化,研究仍停留在实验室阶段。
膜电极(MEA)作为电解池的中心组件,大致可分为两大类:催化剂负载衬底(Catalyst-coated substrate,CCS)和催化剂负载薄膜(Catalyst-coated membrane,CCM)。“衬底”既是电极又是气体扩散层,并且可以采取不同的形式,考虑到成本问题,一般采用不锈钢和镍网电极作为基底。尤其,采用金属泡沫做衬底赋予更大的比表面积,但是这种疏松电极在大电流工作状态下内部会产生大量的气泡,气体的扩散是必须考虑的问题。CCM装置由于其较低的催化剂负载,薄的催化层和疏水性目前广泛应用于PEMWE和碱性燃料电池,此时的气体扩散层用于提供从催化剂层到双极板的电连接,同时也允许所产生的气泡逸出。
通常,全解水电解槽由高活性催化剂的阳极和阴极组成的双电极系统组装成。随后在10 mA·cm-2的电流密度下进行电池电压和长时间稳定性测试。图11a,b是AEMWE电解槽的部件组装示意图,水通过阳极循环,通过得到两个电子产生H+和OH-。然后,OH-通过AEM扩散到阳极侧,通过失去电子形成氧气。通过电解槽复杂的内部组件结构不难看出许多参数会影响AEM电解槽的性能,包括膜的类型和导电性、膜电极处理方法、气体扩散电极的制备、离子体类型、油墨催化剂配比、组装顺序以及进料方法等。PEM电解槽(图11c),IrO2/C和Pt/C分别是经典的阳极和阴极材料,Nafion膜被用作固体电解质,在阴极和阳极之间传导H+,并分离产生的氢气和氧气。

图11 (a)一个AEM水电解系统的横截面示意图;
(b)电解槽组件;
(c)PEM电解槽平面示意图;
(d)无膜流动电解槽的组件和组装结构示意图148Fig. 11 (a)Schematic cross section of an AEM water electrolysis system; (b)AEM electrolyzer components;(c)plane diagram of PEM electrolyzer; (d)the components and assembly of the membrane-free flow electrolyzer 148.
因此,研发催化剂的思路历程要求首先从材料的组成、结构、形貌和性能方面设计出高导电、超疏气/亲水电极材料。随后,该电极材料需要在实验室规模运行中显示出杰出的全解水活性及稳定性。最后,需要从温度、压力、气泡聚集以及传质等方面进一步考虑,才能投入工业规模应用。
膜作为工业电解槽的重要组件,其类型及组装方式的变化决定了流动电解槽的发展。近年来,聚合物膜电解槽已被强调为下一代候选产品。特别是基于PEM的膜占主导地位,允许在高电流密度下运行(> 1 A·cm-2),同时提供加压的高纯度H2(> 99%)。然而,只有贵金属催化剂才能在该电解槽的高腐蚀环境中生存。此外,必须用去离子水来减少膜的降解。因此,高成本是阻碍PEM电解槽大规模应用的一个突出障碍。AEM电解槽由于膜材料成本低,在这种情况下似乎更可取。问题是,目前最先进的AEM的耐久性通常只有~1000 h,而且还未进行产业化开发。因此,设计经济、高效、稳定运行的电解槽仍是未来电解水发展的重点攻克方向。近年,Yan等148报道了一种在高电流密度下使用储量丰富的廉价催化剂进行水分解的无膜流动电解槽。如图11d所示,这种无膜流动电解槽具有三明治状的结构和循环操作模式,用于解耦的整体水分裂。该电解槽由两个物理分离的隔室组成,通过连接线进行电子转移过程,两个隔间具有相同的三明治状结构,包括端板、集流器、双极板、一个130 μm厚的多孔分离器和一个辅助电极(AE)。阳极液(富含O2的1.0 mol·L-1氢氧化钾溶液)和阴极液(富含H2的1.0 mol·L-1氢氧化钾溶液)在反应过程中连续流向指定的隔室。由于这种紧密排列的夹层结构,离子的传输路径要短得多,该无膜电池的欧姆电阻可与质子交换膜相当。并且由于两个反应室的分离,在大电流密度条件下工作所产生大量气泡和过槽压可以明显改善。然而,不难发现该装置无法进行传质,意味着催化剂材料在不断消耗,大大影响了整体的运营成本。因此,他们通过进一步以FeP-CoP/NC双功能催化剂为工作电极,NiOOH/Ni(OH)2为辅助电极进行电解水循环,流程如下:左室为阴极,生成OH-阴离子,通过分离器,在AE处与Ni(OH)2反应形成NiOOH,产生的H2气体通过一个三通阀收集在H2罐中。同时,另一室的工作电极利用NiOOH转化为Ni(OH)2所提供的OH-离子催化阳极液中的OER。形成的氧气也会通过另一个三通阀进入O2罐。当AE材料几乎全部转换时,通过切换电池的电流极性,保证了系统的连续运行。在接下来的循环中,随着左腔室成为阳极,阳极事先被富含O2的阳极所取代,以此保证电解槽的长期运行流动性。但是该装置仍然面临很多问题,无论是组件复杂、装置冗杂、以及转换不变,需要实时监测、反应室气体残留以及长期运行所面临的种种问题都需要进一步优化该操作装置。
5.2 工业电解槽关键问题
面对工业电解槽的要求(图12),首先,需要在大规模合成的前提下设计构建亲水疏气的三维电极材料(图12a),在满足工业需求的同时尽可能延长催化剂的寿命和效率;
其次,结合AEMWE和PEMWE电解槽的优点(图12b),可以以低成本开发在环境下运行高电流密度和效率的电解槽。迈向这些“先进的碱性电解槽”的重要一步是采用基于零间隙概念的电池设计(图12c)149。1976年,Costa和Grimes首次提出零间隙碱性电解池的设计。在碱性电解过程中,零间隙电池设计的工作原理是压缩氢氧根离子导电膜或气体分离器两侧的两个多孔电极,这实现了两个电极之间的间隙等于膜的厚度(< 0.5 mm),而不是传统设置的(> 2 mm),从而显著降低了欧姆电阻。并且,气体扩散层提供了从多孔电极到双极板的电连接,同时允许注入电解溶液,并去除气体产物。使用多孔电极压缩到膜上的零间隙溶液,迫使气泡从电极的背面释放出来,气泡形成一个较小的信号减少空隙分数(图12d149,大量气泡的产生导致可用OH-运输的溶液的体积将大大减少,这种气泡体积称为空隙分数),尽量减少对溶液电阻的影响。在工业电流密度下运行的膜电极产生的大量气泡所造成的实际几何面积减小、槽内阻抗上升、空隙分数的增加以及传质受损等等问题始终阻碍着工业电解槽的进一步发展。尤其,近年来,随着膜电极的迅速发展(图12e),将催化剂直接涂敷在膜电极上,结合零间隙设计,一定程度上可以最大化碱性电解槽的电池效率,但是,目前鲜少有这方面的报导。

图12 电解槽设计的关键因素149Fig. 12 The critical factors of designing alkaline water cell 149.
5.3 电解槽行业未来发展方向
自1931年日本第一台电解水生成机研制成功以来,电解槽的技术进步是渐进性的。目前,我国碱性电解槽已基本实现国产化,具备大规模推广的产业基础。国内碱性电解槽领先企业包括中国船舶718研究所(碱性电解槽主要分为一体式,分体安装式以及集装箱式)、苏州考格利尔竞力、天津大陆制氢、中电丰业、中国华能等,整体技术已接近国际顶尖水平。2020年新增装机容量达到23.47 MW。并且,自2021年隆基股份成立隆基氢能专研电解水产氢电解槽以来,初期便达到年产量500 MW 100台1000 Nm3·h-1电解水设备,并预计未来五年年产量翻倍。同年,中国华能集团研制出世界单槽产能最大的碱性电解槽组件。目前,国内主流碱性电解槽企业均已具备1000 Nm3·h-1以上大功率电解槽的生产能力,2022年碱性电解槽已披露产能接近11 GW。自此,预示着我国碱性电解槽研发迈入了新征程。因此,产量的扩大及产能的进一步提升是未来的发展方向。
碱性介质中缓慢地OER/HER反应速率,极大地限制了工业型碱性电解槽应用的发展。深入研究碱性介质中OER/HER反应机理,明确碱性反应动力学,是开发高活性和高稳定性电解水催化剂的前提。除此以外,大规模合成催化电极进一步应用于工业生产是催化剂发展的根源。本文首先对广泛认可的碱性OER/HER反应机理进行了分类总结,主要包括:OER的晶格氧机制和吸附氧机制;
HER的HBE理论和双吸附位点理论。随后讨论了在理解催化机制的基础上,设计高活性及高稳定
性电解水催化剂的原则:提高催化剂本征活性以及改善催化剂活性位点密度。其次,讨论了构建工业电极的设计原则,包括:大规模合成技术、三维电极表面合成以及超疏气电极制备。膜以及膜电极组装是工业电解槽的关键部件,我们总结了目前电解槽隔膜的种类及发展以及膜电极常用组成。最后,对电解槽种类以及实际工业应用需要注意的问题进行了深入的分析讨论,以期为进一步设计能在工业指标下长期稳定运行的电解槽提供思路及发展方向。表3展现了近五年来工业型电解水催化剂的性能发展。然而,为了更好地制备各种高活性电极材料,仍然存在一些关键问题和挑战:

表3 近五年碱性介质下工业型催化剂性能介绍(≥ 500 mA·cm-2)Table 3 The performance of industrial catalysts (≥ 500 mA·cm-2)in alkaline media in the past five years.
1)演化机制。由于电催化剂在阳极氧化过程中不可避免的重构现象,对于催化剂真正活性位点的探索络绎不绝。其次,由于OER过程的复杂多变性,催化机制也应不断进化发展,宏观的吸附氧机制以及晶格氧机制已经不足以解释OER过程在不同环境下所面对的复杂情况,例如:Sun等81通过温控实验和DFT计算进一步确定,与分子间WNA途径相比,IHNA途径需要的活化能要低得多,进一步明确了当碱性电解液变为中性电解液时,过电位、Tafel斜率和TOFs等显著性能损失的来源。近年来,随着精细结构表征技术以及原位实验的蓬勃发展,电催化剂的实时演化得以被实时监测,如原位X射线吸收光谱(XAS)、原位Raman、原位FTIR、原位SFG、原位EIS、原位DEMS等。
2)电解槽稳定性。就电解槽发展稳定性而言,AEM电解槽大规模工业化仍有很长的探索实践。PEM电解水技术的电流密度高、电解槽体积小、运行灵活、利于快速变载,与风电、光伏(发电的波动性和随机性较大)具有良好的匹配性,预计未来5-10年仍然是电解水发展的主流趋势。因此,进一步研究高效率以及高稳定性的PEMWE电解槽仍然是目前工业电解水发展的主攻方向。然而,受限于PEM水电解制氢的酸性环境、阳极高电位、良好导电性等要求,贵金属催化剂在未来一段时间内仍然是大规模电解槽的催化剂的主要材料。然而,从降低成本的角度考虑,经济效益高的AEM碱性电解槽实用及产业化是未来工业电解水产氢发展的主要方向。
3)混合电解水(Hybrid water electrolysis,HWE)。就常规电解水而言,它在很大程度上受到阳极OER缓慢动力学的限制。因此,将动力学上有利的阳极反应,如生物衍生的化合物氧化和污染物降解32,150,151,与混合水电解中的HER耦合,不仅解决了生物质回收和污染物排放问题,而且还节省了清洁二次发电产氢的能源成本。
猜你喜欢绿氢电解槽碱性碱性电解槽成本最低石油石化绿色低碳(2022年2期)2023-01-06绿氢已成为未来维护能源安全的重要方向科技中国(2022年10期)2022-10-312025年宁夏可再生能源制氢量达到8万吨以上石油化工应用(2022年11期)2022-02-25我国首个万吨级光伏绿氢项目在新疆开建氯碱工业(2021年12期)2021-12-27绿氢成本须下降50%才能与石油竞争石油石化绿色低碳(2021年1期)2021-01-11电解槽焊接施工中的质量控制江西建材(2018年1期)2018-04-04碱性磷酸酶钙-钴法染色的不同包埋方法比较中国组织化学与细胞化学杂志(2017年1期)2017-06-15碱性土壤有效磷测定的影响因素及其控制浙江农业科学(2016年11期)2016-05-04碱性溶液中铂、钯和金析氧性能比较广州大学学报(自然科学版)(2015年4期)2015-12-23碱性介质中甲醇在PdMo/MWCNT上的电化学氧化电源技术(2015年2期)2015-08-22本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0916/655275.html