【www.zhangdahai.com--其他范文】
聂黎行,康 帅#,鲁 静,戴 忠,于健东,王淑红,刘永利,张亚中,魏 锋*,马双成*
1.中国食品药品检定研究院,北京 100050
2.深圳市药品检验研究院,广东 深圳 518057
3.河北省药品医疗器械检验研究院,河北 石家庄 050227
4.安徽省食品药品检验研究院,安徽 合肥 230051
中医药在中华人民共和国香港特别行政区(以下简称香港特区)具有普遍和悠久的应用传统,香港回归祖国后,在国家政策支持下,中医药行业也焕然一新,在香港特区基层医疗体系中发挥着重要的作用。1999年,为规范和促进中医药发展,香港特区成立中医药管理委员会,负责制定及推行有关中药使用、贸易及生产的规管措施。同年《香港中医药条例》实施,其第549 章收载了500 余种中药。香港特区政府也极其重视中药材的品质及安全,为保障公众健康,并将中药推向国际,需要颁布一套结合香港特区实际情况与国际化趋势的标准[1]。为此,香港特区卫生署于2002年起开展《香港中药材标准》(Hong Kong Chinese Materia Medica Standards,HKCMMS)专项,研究制定常用中药材的参考标准。
HKCMMS 由香港特区卫生署组织制订,是对在香港特区使用的中药材质量及检验方法所做的技术规定[2],其宗旨在于推动中药研究、提供参考标准予业界、保障中药材的安全与质量、促进香港中药行业现代化及国际化、鼓励中药贸易。HKCMMS选取研究品种遵循本地常用、经济效益较高、国际关注的原则,并以列于《香港中医药条例》第549章附表2 的品种为优先。一些在安全和品质方面备受国际关注的中药材也被选作研究对象。
历时20 余年,HKCMMS 从无到有,已出版10册,并仍在继续开展研究,形成了成熟的体系。本文介绍了HKCMMS 的发展概况、组织机构和工作程序,结合笔者对HKCMMS 各论起草的工作实践,分析了标准各项目的研究技术特点,并与《中国药典》2020年版进行了比较,以期为中药质量标准研究提供思路和参考。
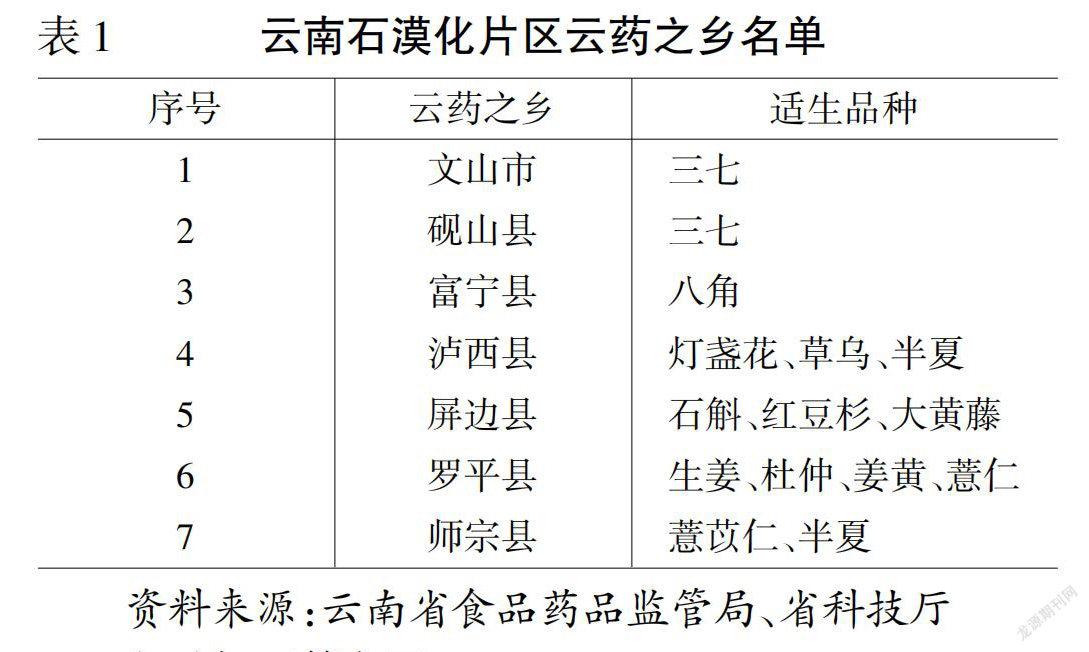
HKCMMS 第1 册于2005年颁布,至2022年已颁布10 册,收录330 种中药材[2-11],见表1。各册均由序言、凡例、专论、附录、索引5 部分组成,以纸质、光盘和电子版形式,中英文双语同时发布,可在香港特区卫生署网站(https://www.cmro.gov.hk/html/gb/useful_information/hkcmms/cmmlist.h tml)搜索、查询和下载。从第1 册起,HKCMMS专论的内容就已基本固定,包括名称、来源、性状、鉴别、检查、浸出物和含量测定项目。除个别品种外,鉴别项一般包括显微、薄层、光谱/色谱指纹图谱鉴别,显微鉴别一般包含横切面和粉末鉴别2 项。检查项一般包括重金属、农药残留、霉菌毒素、二氧化硫、杂质、灰分、水分的检查。性状、显微鉴别、薄层鉴别、光谱/色谱指纹图谱鉴别、含量测定项下附有高清彩色照片或色谱图供参考,薄层鉴别项后还附有化学指标的结构式。

表1 HKCMMS 第1~10 册收录的品种Table 1 Varieties included in volumes 1—10 of HKCMMS
HKCMMS 注重环境保护,方法开发遵循绿色化学原则,避免使用苯、甲苯、三氯甲烷、二氯甲烷等有毒试剂,并鼓励采用超高效液相色谱仪等仪器。另一方面,HKCMMS 密切关注分析学科发展和国内外中药质量评价最新趋势,不断引入新的测试理念和技术,提高标准的专属性、准确性、重现性和可控性。自第2 册起,薄层鉴别项下附有高清彩色薄层图谱,含量测定指标由单一成分迈向多成分测定。自第4 册起,开始引入液相色谱-质谱联用(liquid chromatography-mass spectrometry,LC-MS),采用X-射线衍射(X-ray diffraction,XRD)技术鉴别矿物药材,并附有衍射对照图谱,同时开始使用DNA 检测技术鉴别中药材,并附有凝胶电泳图谱。自第8 册起,含量测定项下附有高清色谱图。自第10册起,引入气相色谱-质谱联用(gas chromatographytandem-MS,GC-MS)技术测定农药残留,并开始全面采用超高效液相色谱(ultra high performance liquid chromatography,UHPLC)技术建立液相色谱指纹图谱的鉴别和含量测定方法。为对部分药材中微生物污染进行有效控制,HKCMMS 在最新的专论研究中引入了水分活度测定。
自统筹阶段起,中国食品药品检定研究院(原中国药品生物制品检定所,以下简称中检院)一直与香港特区卫生署紧密联系,支援HKCMMS 研究计划,并于2001年开始派出中药质量标准研究资深专家赴香港特区指导HKCMMS 的制定。2011年,双方正式签署了合作研究协议,中检院开始作为研究单位正式参与HKCMMS 的研究工作。此后又签订了多次协议,联合地方药检机构共同完成川乌、附子、草乌、马钱子、甘遂、蟾酥、瓜蒌皮、三白草、儿七、洋金花、山银花、肉豆蔻、豆蔻、百部、闹羊花、柏子仁、淫羊藿、山豆根、天仙子(生)、青葙子、千金子、三棱、灵芝、藤黄24 个品种的HKCMMS制定,陆续收录于HKCMMS 第7~9 册。已基本完成的没药、乳香、苏合香、明党参、白芷、藿香、凌霄花和菊花等品种后续也将收入HKCMMS。此外,还完成了黄芩饮片标准先导性研究。
HKCMMS 的组织机构包含决策、管理、评审、研究4 个层面,见图1。香港特区卫生署在中医药规管办公室下设立HKCMMS 组(以下简称“港标办”),专责管理和统筹HKCMMS 研究计划,协调各方运作、定期会商审议。国际专家委员会(International Advisory Board,IAB)对HKCMMS 行使决策权,为HKCMMS 的发展方向、研究及分析方法、内容提供建议,审核及认可研究结果。IAB 主席为香港特区政府卫生署署长,成员包括来自中国(内地和香港)、澳大利亚、泰国、加拿大、美国、日本、德国、英国、奥地利的专家。科学委员会(Scientific Committee,SC)负责审核科研机构提交的研究结果,就实验样品代表性、设计指标科学性、限度合理性等技术性问题提供意见及解决方法。SC由部分国际专家会委员、参与HKCMMS 的科研机构、卫生署及政府化验所代表组成。科研机构是HKCMMS 的主体研究单位,承担收集中药材样品及植物标本、方法研究、草拟标准初稿等工作。目前正式参与HKCMMS 的科研机构包括中检院、香港中文大学、香港浸会大学、香港大学、香港科技大学、香港理工大学、香港城市大学,以及中国台湾的中国医药大学。香港特区政府化验所为HKCMMS 提供技术指导,负责制订重金属、农药残留、黄曲霉毒素的检测方法,并对研究机构的标准草案进行复核。
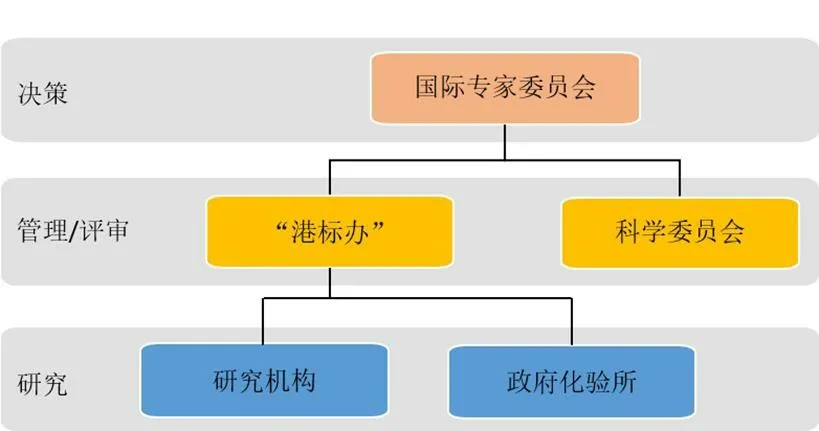
图1 HKCMMS 组织机构Fig.1 Organization of HKCMMS
HKCMMS 的各个参与层面各司其职,形成了一套严密细致的工作程序,见图2。(1)标准投标立项后,研究机构收集药材样品和植物标本,选择化学指标,开展性状、显微鉴别、薄层鉴别、指纹图谱鉴别、检查、浸出物、含量测定等方法研究。在进行重金属、农药残留、黄曲霉毒素检查前,研究机构需测定香港特区政府化验所提供的盲样,通过残留检测能力验证。(2)所有研究项目需按项目计划和港标办安排,分批向SC 汇报,并根据专家意见进行修改。(3)经过SC 审议通过的方法,由香港特区政府化验所进行复核和实验室比对。(4)所有通过实验室比对的品种,由研究机构整合数据和报告,并提交SC 再次讨论,并根据专家意见进行修改。(5)通过SC 审核并经完善的标准初稿,由研究机构向IAB 提交品种综合报告,并根据意见进行修改,形成最终标准。再经港标办编辑委员会进行规范化的整理及文字编篡工作,最终出版发行。SC 会议一般每年举办3~4 次,IAB 会议一般1年至1年半召开1 次。研究机构的所有研究项目必须经SC 会议审核通过后,才能开展后续工作,不可以提前进行。值得一提的是,HKCMMS 的工作语言为英语,各阶段提交审核的材料和进行的汇报均需使用英语。
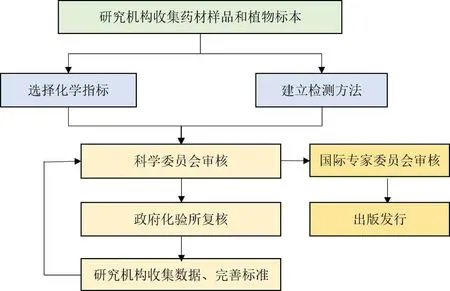
图2 HKCMMS 工作流程Fig.2 Workflow of HKCMMS
3.1 标本和样品收集
标本和样品收集是HKCMMS 研究的基石。研究机构需在研究之初,根据调研情况,初步制定收集计划,通过审核后,方可开展标本和样品收集工作。HKCMMS 研究一般需要10 批以上样品(每个基原物种),其中6 批从不同主产地收集,4 批从香港特区本地市场收集,并保证3 批主产地样品要配套有对应的基原标本,且每个产地不少于5 份。如果在收集的数量上遇到不确定的困难,需提交详细的说明材料。近期研究指南提出,研究机构在采集标本过程中需要提交生境、原植物及反映相关标本采收和制作流程等的图像资料。此外,为减低或避免霉菌毒素的生长,HKCMMS 对标本和样品的储存条件也有明确的要求:温度应在15~20 ℃,湿度应在60%以下。此外,HKCMMS 对研究用标本和样品还有十分严格的记录格式和留样规范。
3.2 化学指标选择
化学指标的选择是HKCMMS 研究的关键环节,需要在方法开发前向SC 汇报,通过审核后,方可开展后续工作。HKCMMS 化学指标的选择考虑专属性、稳定性、生物活性、含量,相应对照品的可获得性和价格等因素。有毒中药材的质量控制指标要包括毒性成分。SC 鼓励研究机构选取2 个或以上的化学指标用于薄层鉴别、指纹图谱鉴别和含量测定,且3 个项目尽量指标一致。应系统查阅品种的研究文献,梳理已报道的被测成分及其检测方法,结合预实验结果确定HKCMMS 的指标成分并购买或制备相应的对照品。薄层鉴别用对照品质量分数应大于95%,指纹图谱鉴别和含量测定用对照品质量分数应大于98%。自第2 册起,HKCMMS每项专论中均在薄层鉴别项最后附有化学指标的名称和结构式(矿物药除外)。在向SC 汇报化学指标的选择时,应详细介绍拟采用对照品的名称、结构式、质量分数及各国药典的使用情况。
3.3 名称和来源
名称包括正名(拉丁药名)、中文名和汉语拼音名;
来源包括原植物的分类类别(包括科、属、种及变种)、药用部位及其状况、采收时间、产地来源、产地加工等有关资料。HKCMMS 名称和来源的制定会着重参考香港特区政府颁布的《中医药条例》第549 章,并保证与《中国药典》2020年版基本一致,同时还重视《中华本草》和最新中文版的《中国植物志》等研究文献的参考。
研究机构须提交有关研究药材名称的文件和文献,有关该药材的原植物鉴定详细研究数据,以及原植物的形态描述、生态环境、生长特性、分布及主要产地等综述资料,选择该药用部位的理由及研究资料,选择该药材最佳采收时间数据及依据等。如名称和来源上有任何混淆及争议,还须进行详细的研究及文献的探讨以查明正确名称及澄清任何混淆。如香港特区政府颁布的《中医药条例》收录有“生附子”,名称、药用部位和产地加工与《中国药典》不一致,经考证,最终确定以“附子(生)”为正名,以“未经炮制的干燥子根”为药用部位,产地加工方式为“晒干”。
3.4 性状
性状主要指药材的外观、颜色、质地、一般内部结构(包括断面特征)、气味/嗅、味道及其他相关数据。HKCMMS 药材性状的描述是对未经处理的样本直接运用感官方法来判断,如眼看(较细小的可借助放大镜)、手摸、鼻闻、口尝。对于多品种来源的药材,若各品种间的性状无明显区别,一般可合并描述;
若性状有明显区别则应分别描述。若情况介乎上述二者之间,则应首先对最重要的来源进行全面论述,然后再分别论述及比较其他来源的药材。
研究机构须对10 批研究样品分别检验2 次,收集20 组数据。测试结果应包括可显示性状特征的照片,而照片应包括以下资料:样本批号、原产地(省)及适当的尺寸比例。如内部结构具有代表性,并有区分药材的鉴别特征,有关特征应拍照,并以独立的编号及标题标示。研究机构通过综合上述研究结果并建议正文内应记载的性状数据的内容。
3.5 鉴别
3.5.1 显微鉴别 显微鉴别是指用显微镜对药材的组织、细胞或其内含物、粉末、解离组织或表面制片的特征进行鉴别的一种方法。HKCMMS 显微鉴别一般包括横切面和粉末2 部分。研究机构须选择具有明显特征的组织、细胞或内含物进行研究。每批样本经制作不少于5 张片子后均能观察的特征才可作为1 批样本的特征。10 批样本均显示相同的特征,有关特征才可作为相应药材的显微鉴别特征。粉末显微特征研究一般以收集通过国家标准6 号筛所得粉末作显微鉴别用。如样本难于粉碎,可用较大的国家标准4 号筛,但必须在方法内清楚列出。研究机构除提供有关该药材显微鉴别的研究资料,还须提供显微鉴别图(图片质量不少于800 万像素,在同一倍数下拍照),并完成整套可显示所有横切面显微鉴别特征的永久性制片。
3.5.2 理化鉴别 鉴于理化鉴别专属性不强,且中药材大多基质复杂,干扰反应,除第4 册的8 个矿物药外,HKCMMS 自第3 册起已不再使用理化手段鉴别中药材。
3.5.3 光谱鉴别 光谱法通过测定所含化学成分与红外、紫外或可见光的相互作用,实现中药材的鉴别。HKCMMS 指南虽规定了光谱鉴别的技术要求,但尚未在专论中使用。
3.5.4 薄层鉴别 薄层鉴别是HKCMMS 使用最普遍的鉴别项目,包括对照品溶液、展开剂、显色剂、供试品溶液、操作程序等内容。供试品溶液制备方法尽量与色谱指纹图谱鉴别项和含量测定项一致,使用2 个或以上的对照品作为对照,供试品与对照品应具有相同的条带和色谱特征,包括比移值(retention factor value,Rf)、斑点颜色及荧光/荧光淬灭等特征。HKCMMS 通常不接受对照药材或对照提取物等作为对照物质,虽然近期研究指南提出,如指标性成分/有效成分尚不明确或得不到,可选用具薄层图谱特征斑点且符合《中国药典》2020年版要求的对照药材作对照,但目前专论仍使用对照品作为对照。固定相均要求使用高效薄层板,为保证斑点显示清晰,要求使用条带点样,不同对照品单独点样,不使用混合对照品溶液。特征条带要求Rf值为0.3~0.7。
在进行薄层鉴别方法的研究和验证时,应首先优化样品提取方法、固定相及展开剂、点样量、预饱和及展开条件、显色方法及检视方法。要求随行溶剂空白进行专属性考察,所有样品均应平行制备2 份供试品溶液分别点样,并要求加标点样。加标点样时,供试品溶液和对照品的点样量均不减半,供试品溶液点样后,所有的对照品溶液按拟定的点样量在同一条带处叠加点样。展开距离应为整数,因此需预先在薄层板上标记溶剂前沿,展至该距离时立即取出。HKCMMS 对不同品牌薄层板引入的方法耐用性未做硬性要求,但需考察不同温、湿度对展开结果的影响,且对多次实验的Rf 值的重现性要求较高(一般绝对偏差不大于0.1)。研究机构应注意需准确记录开发方法时使用的薄层板品牌、温度、湿度,方法一经SC 审核通过,政府化验所采用相同的条件复核方法。研究单位后续如果需要补充实验,也应严格控制相同的条件,因此如在不同季节开展实验,需要使用可调节温湿度的自动展开装置。
研究机构需先向SC 详细阐述方法建立过程中供试品提取溶剂的选择、制备方法的比较,以及固定相、展开剂、显色方法等的比较,溶剂空白、供试品溶液、对照品溶液、加标点样结果,并展示相关的色谱图和Rf 值列表。方法通过审核和复核后,测定10 批样品数据。所有研究完成后向IAB 汇报方法及规定、实验室比对和10 批样品检测结果,并展示相关的色谱图和Rf 值列表。交付资料中除包括仪器和材料、操作方法、色谱条件、方法验证结果,溶剂空白、10 批共20 份供试品溶液、各对照品溶液、加标溶液的高清彩色薄层图谱外,还要求提供各溶液特征条带的Rf 值(精确到小数点后2 位)。
3.5.5 色谱指纹图谱鉴别 色谱指纹图谱鉴别是HKCMMS 的特色项目,包括对照品溶液、供试品溶液、色谱系统、系统适用性要求 [包括指针成分峰的保留时间和峰面积相对标准偏差(relative standard deviation,RSD)、理论板数、与相邻峰的分离度]、操作程序等内容。除矿物药外的所有中药材均要求建立UHPLC 或GC 指纹图谱鉴别方法。根据品种所含化学成分的性质,优化提取方法和分离条件,选择至少4 个分离良好的特征峰用于鉴别,但应排除保留时间在1~3 min(视UHPLC 体积流量情况定)或5 min(GC)前的色谱峰。在所有特征峰中,选定可完全分离并可以通过对照品确定成分的特征峰作为指针成分峰(通常是含量测定指标)。在进行色谱指纹图谱鉴别时,首先采用对照品对供试品中的指针成分峰加以鉴别(要求保留时间RSD<2%),其他特征峰以此指针成分峰作为参考,要求其相对保留时间在规定的可变范围内(一般为±0.03)。HKCMMS 鼓励色谱指纹图谱鉴别项和含量测定项采用相同的色谱条件,因此在方法开发时需要统筹兼顾。除另有规定外,一般使用常规的火焰离子检测器、二极管阵列检测器或蒸发光散射检测器进行检测,如有必要,需采用GC-MS 或UHPLC-MS 技术对特征峰的化学成分加以确认和解析。
在建立色谱指纹图谱鉴别方法时,通过不少于3 批代表性样品对方法加以验证,确定系统适用性要求,包括所有特征峰的分离度、理论塔板数、拖尾因子、半峰宽,以及保留时间和峰面积值的重复性,进而选取1 批样品进行方法学考察,包括专属性(选择另一种对本品种测定无干扰的药材制备空白样品溶液)、精密度、重复性(以所有特征峰的保留时间、峰面积、相对保留时间、相对峰面积的RSD表征,n=5)、加标样品(以样品和加标样品的相对保留时间和相对峰面积表征,n=1)。多来源品种需分品种进行测定。
研究机构需先向SC 详细阐述方法建立过程中供试品提取溶剂的选择、制备方法的比较,色谱柱、流动相、波长等的比较,指针峰和特征峰的确定,以及空白样品溶液、3 批共6 份供试品溶液、混合对照品溶液、加标溶液的色谱图和色谱峰计算结果。方法通过审核和复核后,测定10 批共20 份样品数据。所有研究完成后向IAB 汇报方法及规定、实验室比对和10 批样品检测结果,并展示相关的色谱图和色谱峰计算结果。交付资料中除包括仪器和材料、操作方法、色谱条件和系统适用性要求、方法验证结果,空白溶液、10 批共20 份供试品溶液、对照品溶液、加标溶液的UHPLC/GC 色谱图外,还要求提供各特征峰的保留时间和峰面积、相对保留时间/峰面积(精确到小数点后3 位),以及平行样品上述参数的绝对偏差。如有必要,还需提供特征峰的光谱/质谱图。
3.5.6 XRD 法鉴别 XRD 技术主要用于HKCMMS第4 册中8 个矿物药的指纹图谱鉴别,包括对照品、供试品、系统适用性要求、操作程式等内容。以相应主要无机成分对应的对照品为对照,通过测定对照品和药材粉末的特征衍射角的分布及不同峰强度,获得二者的特征指纹图谱,以对照品的衍射峰的晶面间距(d)及其相对强度数值为特征参数,规定供试品图谱中应检出与对照品一致的5~6 个特征衍射峰,且各峰2θ 值的偏差小于±0.2°。
在建立XRD 鉴别方法时,应根据药材晶体的性质,优化样本的粉碎和处理方法,阳极管类型、X 射线波长、能量、温度、狭缝宽、扫描速度及范围,检测器类型、数据收集方式等测试条件。通过多批次样品的检测确定品种的特征衍射峰及其强度比例,并进行系统适用性(角度的准确性和分离度)、精密度、重复性实验,某些样品还需要开展专属性和耐用性实验。
研究机构需先向SC详细阐述方法的优化过程,拟定特征衍射峰,3 批次以上样品的检测结果。方法通过审核和复核后,测定10 批共20 份样品数据。所有研究完成后向IAB 汇报方法及规定、实验室比对和10 批样品检测结果。交付资料应包括仪器和材料、取样程序、样本制备与测量、XRD 参数、系统适用性要求、方法验证结果、10 批共20 份供试品测试结果及其对应的XRD 图谱。
3.5.7 DNA 鉴别 DNA 鉴别技术目前用于HKCMMS 第7 册川贝母和第10 册蕲蛇的鉴别,均采用聚合酶链反应(polymerase chain reaction,PCR)法,包括所用引物序列、DNA 的提取、实验过程等内容。样品平行操作2 份,并随行阳性提取对照、阴性提取对照和PCR 阴性对照。要求在推荐的测试条件下,样品、平行样品和阳性对照提取物的PCR产物在规定的位置检出相应的DNA 条带,阴性提取对照和PCR 阴性对照不得检出任何条带。
在建立DNA 鉴别方法时,需阐明其科学原理和技术背景,利用物种明确的生物标准物质验证方法的可行性,并采取有效措施预防交叉污染。根据药材的遗传特性选择合适的样本处理、DNA 提取、DNA 扩增、检测扩增物,以及数据分析方法。可采用商品基因提取试剂盒或按权威文献/科研机构发布的方法提取DNA,采用紫外光谱或荧光光谱法测定DNA 提取物的回收率及纯度,以证明方法能够提取足够量的DNA 及有效去除PCR 抑制物。被选取的目标DNA 区域需进行正向和反向DNA 测序,将二者结果重叠后得出准确的共有序列。扩增获得的DNA 条带应不多于1000 个碱基,如果得到2 条或以上DNA 条带,各条带之间长度差距应不少于100 个碱基。建议采用高保真DNA 聚合酶进行PCR,以降低DNA 复制错误的机率,优化退火温度、DNA 聚合酶辅因子浓度、PCR 增效剂,使目标条带产量最大,同时避免非目标条带产生。使用琼脂糖凝胶电泳法检测时,应同时加入DNA 分子量标记物,作为证明电泳结果的有效性和显示PCR 产物的长度和分离度,该DNA 分子量标记物必须包含由100 至1000 碱基的DNA 条带,每个条带应以100 个碱基递增。同时,每个条带应清晰可见及可有效分离。
研究机构需先向SC 详细阐述方法的原理和参数优化过程,3 批次以上样品的检测结果。方法通过审核和复核后,测定10 批共20 份样品数据。所有研究完成后向IAB 汇报方法及规定,10 批样品测定结果。交付资料应包括仪器和材料,样本处理方法,DNA 提取、扩增、检测方法和具体参数,方法验证结果,10 批共20 份供试品测试结果及其对应的原始凝胶电泳图片。在采用琼脂糖凝胶电泳分析时,应详细列出DNA 上样缓冲液、DNA 分子量标记物、琼脂糖凝胶的浓度、电泳缓冲液、DNA 染剂、电泳电压、以及完成电泳时上样缓冲液颜色条带在凝胶上前沿的相对位置等信息。
3.6 检查
HKCMMS 的检查项包括残留检测项 [重金属(铅、镉、砷、汞)、农药残留(9 种有机氯农药)、霉菌毒素(黄曲霉毒素B1、B2、G1、G2)、二氧化硫]和一般检查项(杂质、总灰分、酸不溶性灰分、水分),除矿物药外所有药材都需要检查以上所有项目,每个品种测试10 批药材,每批药材平行操作2 次。
一般检查项按HKCMMS 附录收载方法操作[11],研究机构不需先向SC 提交初步研究结果并等待审核和复核,可直接测试10 批样品。“港标办”根据测定结果按公式(1)计算杂质、总灰分、酸不溶性灰分、水分的理论限度[11]。科研机构对计算出的理论限度与相应品种在其他药典中的相关限度进行比较,选取更严谨的限度值,获得IAB 认可后,确定最终的限度(精确到小数点后1 位)。所有研究完成后向IAB 汇报方法及限度、10 批样品测定结果。交付资料应包括仪器和材料、操作方法、原始数据、10 批共20 份供试品测试结果。
μ为理论限度;
为测定值的平均数;
t为99%置信水平的t值(单尾);
s为测定值的标准偏差;
n为样本的批数;
MU为第1 期HKCMMS 研究时确定的各定量项目的扩展不确定度
HKCMMS 附录[11]收载了所有残留检测项的操作方法和推荐参数,砷、镉、铅、汞采用微波辅助消解-电感耦合等离子体质谱法测定;
9 种有机氯农药残留采用凝胶渗透色谱法固相萃取法净化,GC-电子捕获检测器法测定,GC-MS 验证;
黄曲霉毒素B1、B2、G1、G2 采用免疫亲和柱净化,高效液相色谱分离-柱后碘/光化学衍生-荧光法检测;
二氧化硫采用酸碱滴定法测定。与一般检查项不同,研究机构需先通过各项残留检测能力验证,才能开始正式测定样品中有害残留数据。“港标办”寄送能力验证盲样,研究机构检测后提交检测数据和报告,经“港标办”审核符合要求后,即取得残留检测的能力,可以进行承担品种的样品检测,残留检测能力18 个月内有效。除矿物药或另有规定外,HKCMMS 对所有中药材品种的有害残留规定了统一限度,见表2。研究机构需先向SC 详细阐述最终确定的各残留检测方法和参数。方法通过审核和复核后,测定10批样品共20 份数据。所有研究完成后向IAB 汇报方法、实验室比对和10 批样品检测结果。交付资料应包括仪器和材料、操作方法、10 批共20 份供试品测试结果,空白、对照、样品和方法验证结果及相应的原始图谱。值得指出的是,HKCMMS 不接受有害残留不符合规定的样品纳入研究,故科研机构需在收集样品后进行初筛,保证各项残留检测合格。

表2 HKCMMS 残留限度Table 2 Limits of residues in HKCMMS
3.7 浸出物
与检查项类似,HKCMMS 要求除矿物药外,其余所有药材都须测定水溶性和醇溶性浸出物。药材粉碎后过2 号筛,混匀,以水和70%乙醇测定浸出物。采用3 批样品进行冷浸法与热浸法的比较,选择提取率高,操作简便的方法作为测定方法。特殊品种如有需要,可采用乙醚替代70%乙醇测定醇溶性浸出物含量。浸出物测定方法经SC 审核通过后,不需经过实验室比对,研究机构可直接测试10 批共20 份样品。“港标办”根据测定结果按公式(2)计算浸出物的理论限度[11]。科研机构对计算出的理论限度与相应品种在其他药典中的相关限度进行比较,选取更严谨的限度值,获得IAB 认可后,确定最终的限度(精确到小数点后1 位)。所有研究完成后向IAB汇报方法及限度、10 批样品测定结果。交付资料应包括仪器和材料、操作方法(冷浸法或热浸法)选择依据、各步骤原始称量数据、运算过程、10 批共20 份供试品测试结果。
3.8 含量测定
除极个别品种外,HKCMMS 所有专论均设有含量测定项,大多数品种采用色谱法,矿物药采用滴定法,部分品种没有合适的指标成分,采用比色法或其他方法测定一类成分的总量,如总黄酮、总生物碱、总皂苷、总糖、挥发油等。色谱法采用UHPLC 或GC 技术,主要包括对照品溶液、供试品溶液、色谱系统、系统适用性要求(包括被测成分峰的保留时间和峰面积RSD、理论板数、与相邻峰的分离度)、标准曲线、操作程序、限度等内容。供试品溶液制备方法一般与薄层色谱鉴别项和色谱指纹图谱鉴别项一致,色谱系统、系统适用性要求一般与色谱指纹图谱鉴别项一致。因为含量测定的要求较高,科研机构应先优化确定含量测定项的溶液制备方法和色谱系统,将其用于薄层色谱鉴别项和色谱指纹图谱鉴别项。当被测成分不止1 个,HKCMMS 含量测定项要求配制混合对照品溶液。在建立含量测定方法时,供试品溶液的制备应采用多次、完全提取的方法,HKCMMS 不接受定量加提取溶剂,补足减失质量的平衡提取方法。应对样品经多次提取的供试品溶液进样考察,测定峰的检测情况至该次的提取量可以忽略为止。此外,HKCMMS 的定量方法为标准曲线法,即随行测定系列浓度对照品溶液,制备工作曲线,用工作曲线计算样品含量,不接受外标一点法计算。如采用衍生化方法测定含量,尽量选用衍生后的化合物作为对照品,如购买或制备困难,可选原型成分衍生后进行测定,但需少量制备相应衍生物并进行结构确证,指认色谱峰。
在建立含量测定方法时,通过不少于3 批代表性样品对方法加以验证,确定系统适用性要求,包括所有被测峰的分离度、理论塔板数、拖尾因子、半峰宽,以及保留时间和峰面积值的重复性,进而选取1 批样品进行方法学考察,包括专属性、检出限、定量限、线性、精密度、重复性、回收率。专属性考察所用空白对照溶液,选择另一种对本品种测定无干扰的药材,如川乌的含量测定选择浙贝母、柏子仁的含量测定选择苦杏仁作为空白。如测定成分是普遍存在的非特征性成分,例如,脂肪酸、氨基酸等,难以找到无干扰的药材,可采用适宜溶剂,将拟测定的成分提取后去除,残留药渣作为空白样品,按程序提取制备空白对照溶液。检出限是指在统计学置信区间内,样本中被测物能够检测到的最小量。实际操作时,先估算信噪比3∶1 的加标浓度,在空白样品中加入对照品,依法制得加标溶液,进样实验是否可检出,若由于本底影响不能检出,则适当增加加标浓度。随行工作曲线,3 d 内测定7次(每天至少测定1 次),计算加标溶液浓度,求得7 次测定值的RSD,供试液检出限为该标准偏差的3.14 倍(单位为mg/L),再根据称样质量、制成体积、稀释倍数等计算得到方法检出限(单位为mg/kg)。此后还需向空白样品中准确加入适量对照品,使空白加标溶液浓度为供试液检出限,进样,得到检出限色谱图附于报告。定量限是指在不确定度可接受的区间,样本中被测物可测定的最低量。向空白样品中准确加入适量对照品,使空白加标溶液浓度为供试液检出限的5 倍,随行工作曲线,连续进样5 次,计算含量的RSD 值(要求小于5%)及回收率(要求单个及平均回收率在90%~100%)。如测定结果满足要求,则该加入量对应的含量为方法定量限,单位为mg/kg。如测定结果不能满足要求,则可适当扩大加标浓度(6~10 倍),重新确定定量限。线性一般考察7 个浓度水平,比定量用工作曲线范围更宽,最低点可参考定量限浓度,最高点要涵盖工作曲线,相关系数应不低于0.995。精密度实验取工作曲线中间浓度对照品重复进样5 次,随行标准曲线对照,计算浓度,要求RSD<2%。重复性实验取同一批样品,平行测定5 次,要求含量的RSD<5%。回收率实验在已知含量的样品中加入对照品,平行制备5 份1∶1 加样回收溶液(无需减半加标),测定,一般要求单个回收率和平均回收率均在95%~105%。基于方法的复杂性和含量水平,某些回收率要求可扩展至90%~110%。HKCMMS不要求所有含量测定方法均考察稳定性,但当测试样本在进行检测前须作进一步化学处理,应对此处理的效率进行研究,一般来说,应监测处理过程前后初始物或生成物的含量,以确定处理过程的最佳时间。HKCMMS 也不要求进行耐用性实验,考察不同色谱柱和仪器对测定结果的影响,但对保留时间的偏移容忍度较低,进行方法学考察和样品测定时,尽量固定仪器和色谱柱。报告正文和附件中表格和附图均要求注明保留时间,如不同项目保留时间偏移较大(如RSD 超过2%),可要求重新进行实验。
除方法学验证外,研究机构需制定质量控制方案,并按此方案对检测结果质量进行控制。每次分析前,测定空白溶液以排除污染,空白溶液中被测成分的保留时间处信号响应不得高出方法检出限。标准曲线不得少于5 个浓度水平,相关系数应不低于0.995,否则应在测定样品前重新制备系列工作对照。按照标准曲线中间浓度,另取对照品配制初始校准标准溶液检查工作曲线准确性,测定值和预期值相差应不大于10%。每20 个样品随行测定1 次初始校准标准溶液,测定值和预期值相差应不大于10%。每批样品平行测定,相对平均偏差应不高于10%。每20 个样品测定1 次加标样品,回收率应在90%~110%。如有任何一项质量控制指标达不到要求,应停止检测,纠正问题、校准仪器、重新测定。
研究机构需先向SC 详细阐述方法建立过程中供试品溶液制备提取溶剂和提取次数的选择、制备方法的比较,色谱柱、流动相、波长等的比较,系统适用性、精密度、重复性、回收率考察结果,以及空白样品溶液、3 批共6 份供试品溶液、混合对照品溶液、加标溶液的色谱图和含量测定结果。方法通过审核和复核后,测定10 批共20 份样品数据。“港标办”根据操作步骤、方法学验证和样品测定结果计算不确定度,并按公式(2)计算被测成分的理论限度。科研机构对计算出的理论限度与相应品种在其他药典中的相关限度进行比较,选取更严谨的限度值,获得IAB 认可后,确定最终的限度(保留2 位有效数字)。所有研究完成后向IAB 汇报方法及限度、方法学验证结果、实验室比对和10 批样品测定结果,并展示相关的色谱图。交付资料包括仪器和材料,操作方法,色谱条件和系统适用性要求,方法验证结果,空白溶液、供试品溶液、对照品溶液、加标溶液的UHPLC/GC 色谱图,10 批共20 份供试品的测定结果及每批样品含量的平均值,各被测峰的保留时间和峰面积,质量控制参数测定结果。
HKCMMS 与《中国药典》2020年版一部均收载了大量中药材各论,但二者在项目设置、正文内容、方法开发、方法验证、限度拟定、工作程序等方面各有特色,值得相互借鉴。
4.1 项目设置
《中国药典》中药材品种各论可分别列有品名、来源、性状、鉴别(包括横切面显微鉴别、粉末显微鉴别、薄层鉴别、DNA 鉴别)、检查(包括杂质、总灰分、酸不溶性灰分、水分、重金属及有害元素、农药残留、黄曲霉毒素、内源性有害物质等)、浸出物(包括水溶性和醇溶性浸出物)、特征图谱、含量测定、炮制、性味与归经、功能与主治、用法与用量、注意、贮藏等项目。其中品名、来源、性状、性味与归经、功能与主治、用法与用量、注意是所有药材各论均列有的项目,并自《中国药典》2020年版起大幅度增加了炮制方法及相应饮片标准,充分体现了中医药特点,为临床用药提供理论指导。各项鉴别、检查、2 种浸出物、含量测定项根据品种具体情况和当前研究水平有选择性地列入,仅个别品种收录了特征图谱检测项。此外,对禁用农药和二氧化硫残留进行严格控制,《中国药典》2020年版四部“0212 药材和饮片检定通则”中规定:“药材及饮片(矿物类除外)的二氧化硫残留量不得过150 mg/kg;
药材及饮片(植物类)33 种禁用农药不得检出(不得过定量限)”,并根据具体风险情况在各论中规定了部分品种的其他有机氯类农药、重金属和有害元素(铅、镉、砷、汞、铜)或黄曲霉毒素限度[12]。HKCMMS 各品种专论项目高度统一,除极个别品种外,均列有名称、来源、性状、鉴别、检查、浸出物、含量测定项,毒性药材还列有警告项。大多数非矿物药品种均规定了横切面显微鉴别、粉末显微鉴别、薄层鉴别、色谱指纹图谱鉴别项,个别化学指标不易获得的品种采用DNA 鉴别代替薄层鉴别和/或色谱指纹图谱鉴别。另一方面,均要求测定杂质、总灰分、酸不溶性灰分、水分、重金属(铅、镉、砷、汞)、农药残留(9 种有机氯农药)、霉菌毒素(黄曲霉毒素B1、B2、G1、G2)、二氧化硫、水溶性和醇溶性浸出物,并对有害残留限度做出了统一规定。所有矿物药品种均规定了粉末显微鉴别、理化鉴别、XRD 鉴别、含量测定项。总体而言,与《中国药典》相比,HKCMMS 的项目设置更加全面,尤其横切面显微鉴别、粉末显微鉴别、薄层鉴别、浸出物测定的广泛应用,以及矿物药的XRD 鉴别和含量测定项的普遍设置,提高了质量标准的可控性。需要指出的是,HKCMMS 将色谱指纹图谱列入鉴别项与《中国药典》将指纹图谱或特征图谱单列不同,但从操作方法看,HKCMMS 的指纹图谱与《中国药典》的特征图谱更加类似。安全性方面,HKCMMS 的项目设置和限度与《中国药典》各有异同,如二者对二氧化硫残留的规定一致,《中国药典》除对33 种禁用农药和二氧化硫做出统一规定外,其余检查项,多基于具体品种的实际情况进行有针对性地控制,并在2020年版提出了基于风险评估的“中药有害残留物限量制定指导原则”[12],而HKCMMS 则对所有品种的一般检查项设置和残留检查项设置和限度采取“一刀切”的控制思路。
4.2 正文内容
虽然HKCMMS 收录的中药材品种数量远不及《中国药典》,但各论正文表述更加全面和详尽,除一般检查和有害残留检查照附录操作外,其余各个检测项目均详细注明了试剂种类、溶液配制方法、操作程序,对于有毒的试剂和危险的操作,还会在正文中提出警示。性状、横切面显微鉴别、粉末显微鉴别、薄层鉴别、色谱指纹图谱鉴别、DNA 鉴别、XRD 鉴别、含量测定等项目,均配有高清彩色图片供标准使用人员参考。所有标准均以中英文2 种形式同时发布,并可在网站免费、方便地查阅、下载。HKCMMS 的以上特点,即使放之世界各国/地区药典范围内,仍具有先进性和参考意义。
4.3 方法开发、方法验证和限度拟定
除项目设置和正文内容外,HKCMMS 在方法开发、方法验证和限度拟定方面的技术特点也与《中国药典》有所区别。名称方面,HKCMMS 在与《中国药典》基本保持一致的前提下,会着重参考香港特区政府颁布的《中医药条例》第549 章。来源方面,HKCMMS 对基原学名及来源科属名称的确定会更多考虑国际植物分类最新研究成果,对产地加工方法会较多考虑调研实际结果,并趋向于更加科学和明确的表述方式。标本和样品收集方面,除样品批次要求与《中国药典》(不少于15 批)有所差异外,对样品和标本的记录格式和留样规范,以及标本采集和制作过程的配套图像数据都有详细的要求。性状和显微鉴别方面,HKCMMS 与《中国药典》技术要点基本一致,但对于有毒药材,考虑检验实际,性状描述中会略去“口尝”特征;
HKCMMS正文中会附上典型的药材大样、表面观、断面等性状鉴别特征图、横切面墨线图和组织详图,以及粉末特征各特征组合图以供参考。化学分析方面,HKCMMS 与《中国药典》在技术方面最显著的区别是前者不使用对照药材作为标准物质,且要求薄层鉴别、色谱指纹图谱鉴别、含量测定尽量采用相同的对照品和供试品提取方法,而后者薄层鉴别项供试品提取方法一般与含量测定项不同,且大规模使用经济的对照药材,体现了中药的整体性,部分品种还针对不同极性成分建立了多个薄层鉴别方法,信息更加丰富,可以更全面地反映中药材的内在质量[13-14]。《中国药典》在指标成分的选择方面会更多地考虑质量相关性,并不过于倚重单体成分的活性和可获得性。质量标志物理念[15-22]也可为《中国药典》中药材标准指标选择提供科学思路。HKCMMS 对仪器和材料要求较高,薄层鉴别采用高效薄层板,不接受普通薄层板,自第10 册起色谱指纹图谱鉴别和含量测定采用超高效液相色谱,不接受高效液相色谱,上述要求可在一定程度上提高方法的准确性、重复性和重现性,而《中国药典》则会更多地考虑方法的经济成本和适用性。此外,HKCMMS 严格禁用甲苯、苯、三氯甲烷、二氯甲烷等毒性试剂,在开展各个化学分析项目时需要注意。方法学验证方面,HKCMMS 薄层鉴别和色谱指纹图谱鉴别研究均需制备平行样品和加标样品。含量测定采用标准曲线法定量,不接受《中国药典》普遍采用的外标一点法,供试品溶液的制备采用“完全提取法”,不接受《中国药典》普遍采用的“平衡提取法”。方法学验证方面,HKCMMS 色谱指纹图谱鉴别和含量测定专属性考察用空白样品不是试剂空白,而是另一种对测定无干扰的中药材;
检出限、定量限的测定难度较大、耗时较长;
精密度、重复性、回收率均采用n=5 的方式测定,精密度以测定浓度偏差而非峰面积偏差表示;
回收率加标采用1∶1 的形式而非样品减半加标。与《中国药典》不同,HKCMMS 对方法的稳定性考察未做强制性要求,也不要求考察耐用性,但对薄层色谱的Rf 值和UHPLC/GC 色谱的保留时间偏移容忍度低。HKCMMS 对含量测定过程的质量控制提出了明确要求,每次分析前需随行空白溶液以保证系统无干扰,每20 个样品随行测定1 次初始校准标准溶液和加标样品,以保证工作曲线和样品测定结果的准确性。HKCMMS 的杂质、总灰分、酸不溶性灰分、水分检查、浸出物、含量测定等项目的限度拟定引入了不确定度,提高了标准的科学性,但《中国药典》对标准制修订采用的中药材样品批数要求更多,可以更全面、准确地反映品种的质量现状。
4.4 工作程序
与《中国药典》“药典委员会立项-研究单位起草-复核单位复核-专业委员会审议-标准公示-专业委员会审核-国家药品监督管理局颁布”的工作流程不同,研究机构在HKCMMS 的化学指标选择、方法建立和验证、数据收集等各个研究阶段均需及时向SC 和/或IAB 进行书面和英文口头汇报。二者工作程序最大的区别是《中国药典》立项后开始研究,所有项目研究完后再提交复核单位复核,复核完成后再向国家药典委员会提交资料进行审核,而HKCMMS 则分阶段进行严格管理。残留检查项目还需通过“港标办”组织的能力验证后,才可开展正式样品测定。标准复核方面,《中国药典》制修订的所有项目均需经复核单位复核,但对定量结果的重现性并未做出明确规定。HKCMMS 则是以政府化验所实验室比对的形式对除一般检查项和浸出物以外的项目进行复核,并专门制定了实验室间对比测试结果的评估指引,规定了评估实验室间对比测试结果的统计学方法。“港标办”对所有研究机构实施严格的项目管理,并保持密切沟通,明确规定了各研究阶段成果的交付时间,并规范了报告的具体细节。总体来讲,HKCMMS 的起草工作更加细致、繁琐,对于习惯《中国药典》标准制修订工作的研究人员而言,有很多需要适应的工作程序和技术细节。
HKCMMS 研究思路清晰、程序严谨,注重基原调查,采集、制作植物标本,尊重药材的现时使用品种和使用状况。通过20年的研究,收藏、积累了大量可靠的原植物标本和药材样本,丰富和拓展了中药材的质量控制指标。中检院通过HKCMMS 的合作研究,更加理解和掌握了HKCMMS 制定的各项原则和技术特点,香港特区卫生署在标准研究和制定方面也更加需要和重视内地研究机构和专家的意见和建议。中检院协助香港特区卫生署开展HKCMMS 项目的工作,将有力加强中国内地与香港特区在中药标准研究方面的紧密合作,促进双方的合作与交流。今后还将不断拓展合作领域,将技术交流、人才培养和深化合作作为今后2 地中药检测和标准研究领域主要合作事项,提升研究人员的科研素质和国际交流水平。相信在各方共同努力下,HKCMMS 会继续发挥其作用,让患者可安心使用以实证为基础、安全和高素质的中药,对推动中药材质量标准的提高,进而与国际标准接轨,保障全球公众健康,以及促进中药贸易做出应有的贡献。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢药典图谱供试绘一张成长图谱少先队活动(2020年12期)2021-01-14不同处方配比及提取工艺对银柴退热汤解热镇痛抗菌抗炎作用的影响山西中医药大学学报(2020年2期)2020-06-062015年版《中国药典》脑安胶囊项下阿魏酸供试品溶液制备方法的改进中成药(2018年12期)2018-12-29补肾强身片UPLC指纹图谱中成药(2017年3期)2017-05-1766种中药材进入欧洲药典 未来目标达到300种人民周刊(2016年11期)2016-06-30主动对接你思维的知识图谱领导科学论坛(2016年9期)2016-06-052015年版《中国兽药典》编制完成中国猪业(2016年1期)2016-01-28Flexible ureteroscopy:Technological advancements,current indications and outcomes in the treatment of urolithiasisAsian Journal of Urology(2015年3期)2015-12-16《中国药典》2010版毒性中药分析中国中医药现代远程教育(2014年11期)2014-08-08国家药典委员会对《中国药典》2015年版三部增修订内容征求意见中国医药生物技术(2014年4期)2014-01-23本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0919/656317.html