【www.zhangdahai.com--其他范文】
陈俊沲,余 平,杨佳力,万浩芳,张双双,石森林*
(1.浙江中医药大学药学院,浙江 杭州 310053;
2.浙江省立同德医院,浙江 杭州 310012)
大黄始载于《神农本草经》,为蓼科植物掌叶大黄Rheum palmatumL.、唐古特大黄Rheum tauguticumMaxim.ex Balf.或药用大黄Rheum offciualeBaill.的干燥根及根茎,具有清热泻火、活血祛瘀的作用[1],其主要活性成分为蒽醌类衍生物,分为游离型蒽醌与结合型蒽醌,其中前者包括大黄素、芦荟大黄素、大黄酚、大黄素甲醚、大黄酸等,具有较好的抗炎、抗肿瘤作用[2-3]。但大黄蒽醌的水溶性差,口服生物利用度低,从而限制了该成分应用[4-5],目前主要通过将其制成脂质体、聚合物胶束、固体脂质纳米粒等制剂来改善在水中的溶解度,但存在工艺复杂、制备过程中会引入有毒试剂等问题[6]。
包合物是一种工艺简单、安全可靠的制剂,其中羟丙基-β-环糊精具有很好的水溶性[7-9],将药物包合于其中能显著增加其溶解度,提高生物利用度,但它受诸多因素的影响,仅根据单一指标难以筛选出最优制备工艺。层次分析法是一种将复杂问题中的各个因素划分为相互联系的有序层次,再确定权重系数的方法[10-11],适用于大黄蒽醌包合物制备工艺的研究。本实验优化大黄蒽醌羟丙基-β-环糊精包合工艺,以增加其在水中的溶解度,以期为大黄蒽醌的制剂研究提供理论依据。
1.1 仪器 Nicolet IS50 红外分光光度计,购自美国赛默飞世尔科技公司;
ZF-I 型三用紫外分析仪,购自上海顾村电光仪器厂;
S-3000N、E-1010 扫描电子显微镜,购自日本日立公司;
XRD-6100 X 射线衍射仪,购自日本岛津公司;
Agilent1200 series 高效液相色谱仪,购自美国Agilent 公司;
CP313 电子天平,购自奥豪斯仪器(上海)有限公司;
XS105 电子分析天平,购自梅特勒-托利多仪器(上海)有限公司;
KH-250DB 数控超声波清洗器,购自昆山禾创超声仪器有限公司;
SHB-III 循环水式多用真空泵,购自郑州长城科工贸有限公司;
DF-101S 集热式磁力搅拌器,购自巩义市予华仪器有限公司;
RE-3000 旋转蒸发仪,购自上海亚荣生化仪器有限公司;
DNG-5070A 电热恒温鼓风干燥箱,购自上海精宏实验设备有限公司。
1.2 试剂与药物 大黄蒽醌原料药(实验室自制,纯度>50%)。大黄素(批号110756-200110)、大黄酚(批号110796-200716)对照品,购自中国食品药品检定研究院;
大黄酸(批号 T20A8F42628)、芦荟大黄素(批号T28D6F8264)、大黄素甲醚(批号T26A8F34784)对照品及羟丙基-β-环糊精(批号Z13N11Y130870),购自上海源叶生物科技有限公司。乙酸镁(批号080101),购自上海泗崟化工有限公司。甲醇(批号20210605)、无水乙醇(批号20200811)为色谱纯,购自广东光华科技股份有限公司;
其他试剂均为分析纯;
水为超纯水。
2.1 包合物制备 按1∶40 比例称取适量大黄蒽醌与羟丙基-β-环糊精,分别用无水乙醇、纯化水完全溶解,置于磁力加热搅拌器上,包合一定时间后静置,在50 ℃下旋转蒸发除去乙醇,0.45 μm 微孔滤膜过滤,置于50 ℃水浴锅上蒸干后移到50 ℃真空干燥箱中干燥,即得。
2.2 包合率、得率测定 精密称取适量包合物,甲醇溶解,超声处理30 min,混匀后定容至10 mL,0.22 μm 微孔滤膜过滤,分别采用课题组前期建立的HPLC 法、醋酸镁-甲醇溶液显色法测定峰面积、吸光度,计算包合率、得率,公式分别为
2.3 权重系数确定 在制备包合物时,需同时考虑包合率、得率等指标,由于大黄蒽醌包含多个游离型蒽醌,故选择任何1 个单一指标均无法得到最优制备工艺,但同时选择多个指标时又会产生冲突。因此,本实验基于评价包合工艺的关键指标,以及各游离蒽醌药效活性与含量等综合因素之间的关系[12-14],以芦荟大黄素、大黄素、大黄酚、大黄素甲醚、大黄总蒽醌包合率、包合物得率为权重指标并予以量化,再通过YAAHP 软件中的1~9 标度法对其进行两两比较以评判重要性,构建成对比较的判断优先矩阵,赋予各项指标间的相对评分[15-16],具体见表1~2。

表1 各指标评分标准
根据表2 结果,采用层次分析法计算各指标权重系数,结果见表3。随机一致性比率CR =CI/RI,当CR<0.1 时,判断矩阵具有满意的一致性及权重计算正确性[17-19],表3显示CR 为0.068 0,表明权重系数有效。
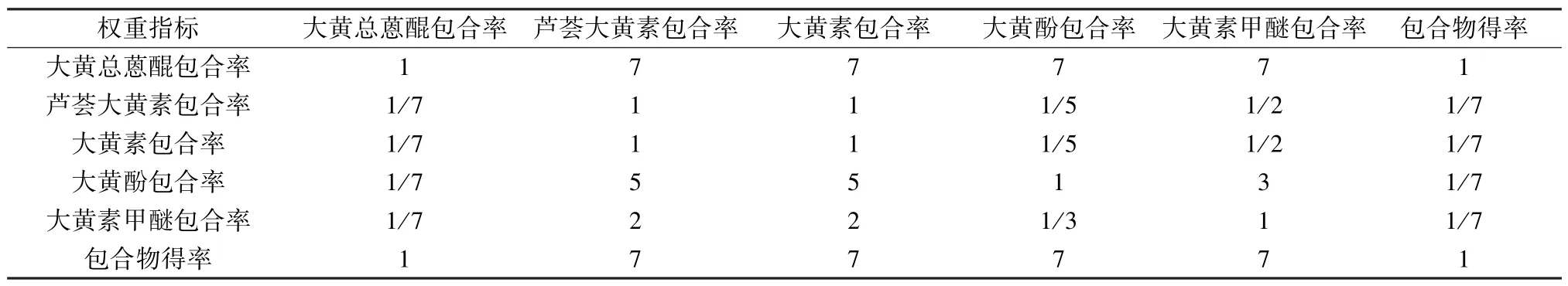
表2 各指标成对比较的优先判断矩阵

表3 各指标权重系数
最后计算综合评分,公式为综合评分=(芦荟大黄素包合率/最大值×0.038 5+大黄素包合率/最大值×0.038 5+大黄酚包合率/最大值×0.115 4+大黄素甲醚包合率/最大值×0.053 4+大黄总蒽醌包合率/最大值×0.379 9+包合物得率/最大值×0.379 9)×100。
2.4 单因素试验
2.4.1 大黄蒽醌与羟丙基-β-环糊精比例 分别按1∶10、1∶20、1∶40 比例称取适量大黄蒽醌与羟丙基-β-环糊精,按“2.1” 项下方法制备包合物,按“2.2” 项下方法测定包合率、得率,结果见表4。由此可知,当大黄蒽醌与羟丙基-β-环糊精比例为1∶40 时,包合率、得率、综合评分较高,可能是由于包合物形成主要依靠羟丙基-β-环糊精与药物的包合作用,而前者越大可使后者被包合的量也越多,但考虑到成本、辅料用量等问题,两者比例并非越大越好。因此,选择大黄蒽醌与羟丙基-β-环糊精比例为1∶40。

表4 大黄蒽醌与羟丙基-β-环糊精比例对包合率、得率的影响(n=3)
2.4.2 包合温度 分别在包合温度30、40、50、60 ℃下按“2.1” 项下方法制备包合物,按“2.2” 项下方法测定包合率、得率,结果见表5。由此可知,当包合温度为60 ℃时,包合率降低,导致综合评分呈下降趋势,不利于包合物制备,可能与大黄蒽醌热稳定性有关。因此选择包合温度为30~50 ℃。

表5 包合温度对包合率、得率的影响(n=3)
2.4.3 搅拌速度 分别在搅拌速度100、200、400、600 r/min 下按 “2.1” 项下方法制备包合物,按“2.2” 项下方法测定包合率、得率,结果见表6。由此可知,随着搅拌速度增加包合率、得率、综合评分不断升高,但在600 r/min 时综合评分反而降低,可能是由于搅拌速度过快导致药物无法进入羟丙基-β-环糊精的空穴结构中所致。因此,选择搅拌速度为100~400 r/min。

表6 搅拌速度对包合率、得率的影响(n=3)
2.4.4 包合时间 分别在包合时间1、2、4、6 h 时按“2.1” 项下方法制备包合物,按“2.2” 项下方法测定包合率、得率,结果见表7。由此可知,随着包合时间延长包合率、得率、综合评分不断升高,但在6 h 时综合评分反而降低,可能与大黄蒽醌在溶剂中的稳定性有关,并且时间过长成本也会较大。因此,选择包合时间为1~4 h。

表7 包合时间对包合率、得率的影响(n=3)
2.5 正交试验 在单因素试验基础上,选择搅拌速度(A)、包合温度(B)、包合时间(C)作为影响因素,综合评分作为评价指标,采用L9(34)正交表进行试验,因素水平见表8,结果见表9。

表8 因素水平
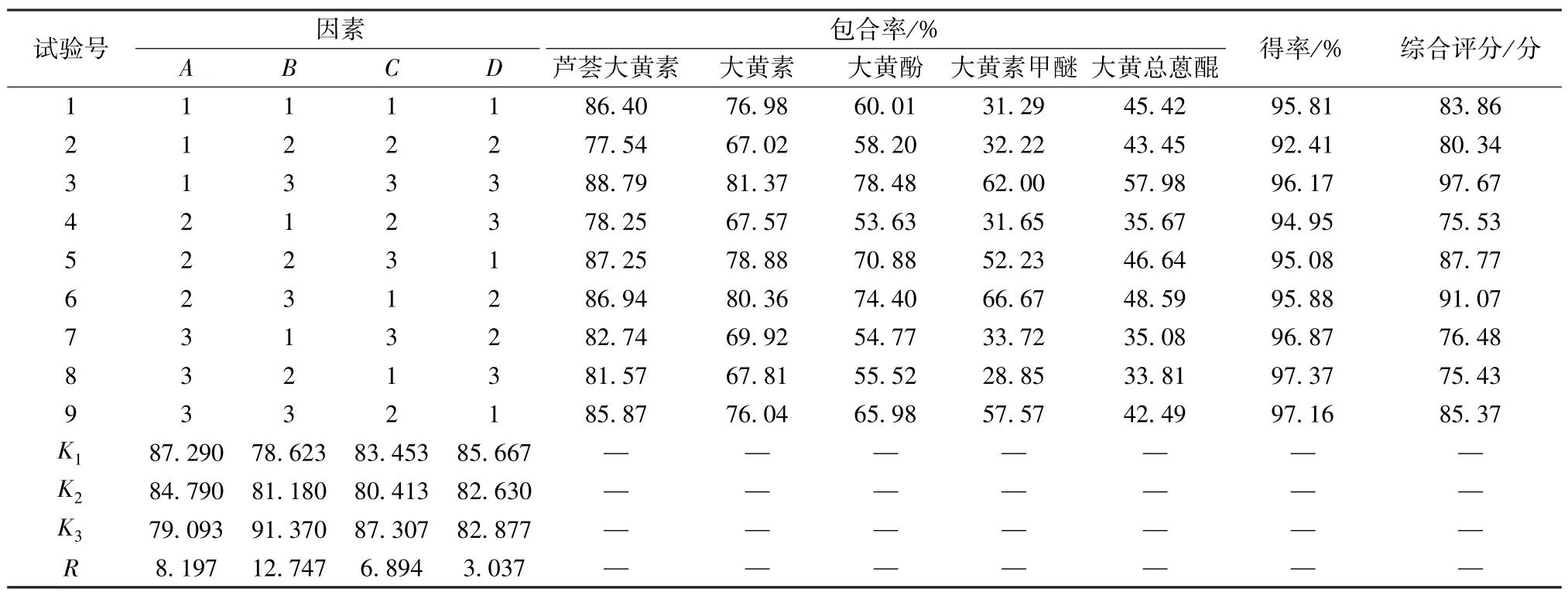
表9 试验设计与结果
方差分析见表10,可知各因素影响程度依次为包合温度>搅拌速度>包合时间。最终确定,最优工艺为A1B3C3,即搅拌速度100 r/min,包合温度50 ℃,包合时间4 h。

表10 方差分析
按上述优化工艺进行验证试验,即按1∶40 比例称取大黄蒽醌与羟丙基-β-环糊精,分别用无水乙醇、纯化水完全溶解,控制包合温度为50 ℃,搅拌速度为100 r/min,包合时间为4 h,按“2.1” 项下方法制备包合物,按“2.2”项下方法测定包合率、得率,平行3 次,结果见表11。由此可知,该工艺稳定可行,可用于制备大黄蒽醌羟丙基-β-环糊精包合物。

表11 验证试验结果(n=3)
2.6 包合物表征
2.6.1 红外光谱 取原料药、羟丙基-β-环糊精、物理混合物、包合物适量,采用KBr 压片法,设定分辨率为4 cm-1,在4 000~500 cm-1波数处进行分析,结果见图1。由此可知,包合后1 629.17 cm-1等波数处原料药吸收峰已经消失或明显减弱,表明药物进入羟丙基-β-环糊精空腔,其红外振动受到限制,包合物成功形成;
包合物特征峰与羟丙基-β-环糊精相似,峰形相同;
物理混合物红外图谱是由吸收较强的羟丙基-β-环糊精红外图谱与吸收较弱的原料药红外图谱加和而成,并且与羟丙基-β-环糊精相似,可能是由于物理混合物中也含有大量该辅料所致。
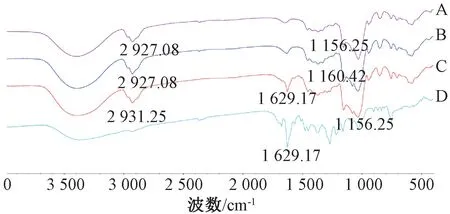
图1 各样品红外光谱图
2.6.2 X 射线衍射 设定条件为Cu 靶(40 kV,40 mA);
步进扫描0.02°/步;
扫描范围10°~80°;
扫描速度2°/min,对羟丙基-β-环糊精、原料药、物理混合物、包合物进行分析,结果见图2。由此可知,羟丙基-β-环糊精、原料药在10°~35°处有多个强结晶特征衍射峰,后者衍射峰与前者差异较大,表明原料药已被成功包合入羟丙基-β-环糊精的空穴中。

图2 各样品X 射线衍射图
2.6.3 扫描电镜 取羟丙基-β-环糊精、原料药、物理混合物、包合物适量,经导电胶粘样、喷金镀膜后固定于铜板上,在扫描电镜下观察其外观,结果见图3。由此可知,羟丙基-β-环糊精呈中空圆球状;
原料药呈表面粗糙的巨大团块;
物理混合物为中空圆球与粗糙团块的简单混合物;
包合物为表面光滑的细小颗粒,表明其成功形成。

图3 各样品扫描电镜图
2.7 平衡溶解度测定 取5 mL 纯化水至离心管中,加入过量包合物,置于恒温振荡器中,温度保持(25±1)℃,振摇24 h,7 000 r/min 离心10 min,取上清液,过0.22 μm微孔滤膜,稀释适当倍数,进行HPLC 分析,取适量上清液,采用醋酸镁-甲醇溶液显色法测定大黄总蒽醌含量,结果见图4。由此可知,未经包合的原料药饱和水溶液中未检出芦荟大黄素、大黄素、大黄酚、大黄素甲醚,大黄总蒽醌溶解度仅为(69.25±4.10)μg/mL;
包合后,上述4 种成分溶解度分别为(166.78±22.50)、(117.40±11.14)、(31.17±5.71)、(11.08±3.07)μg/mL,出现了明显色谱峰,大黄总蒽醌溶解度达(399.24±42.93)μg/mL,与包合前相比增加了5.77 倍,表明羟丙基-β-环糊精包合物能有效改善大黄蒽醌水溶性。
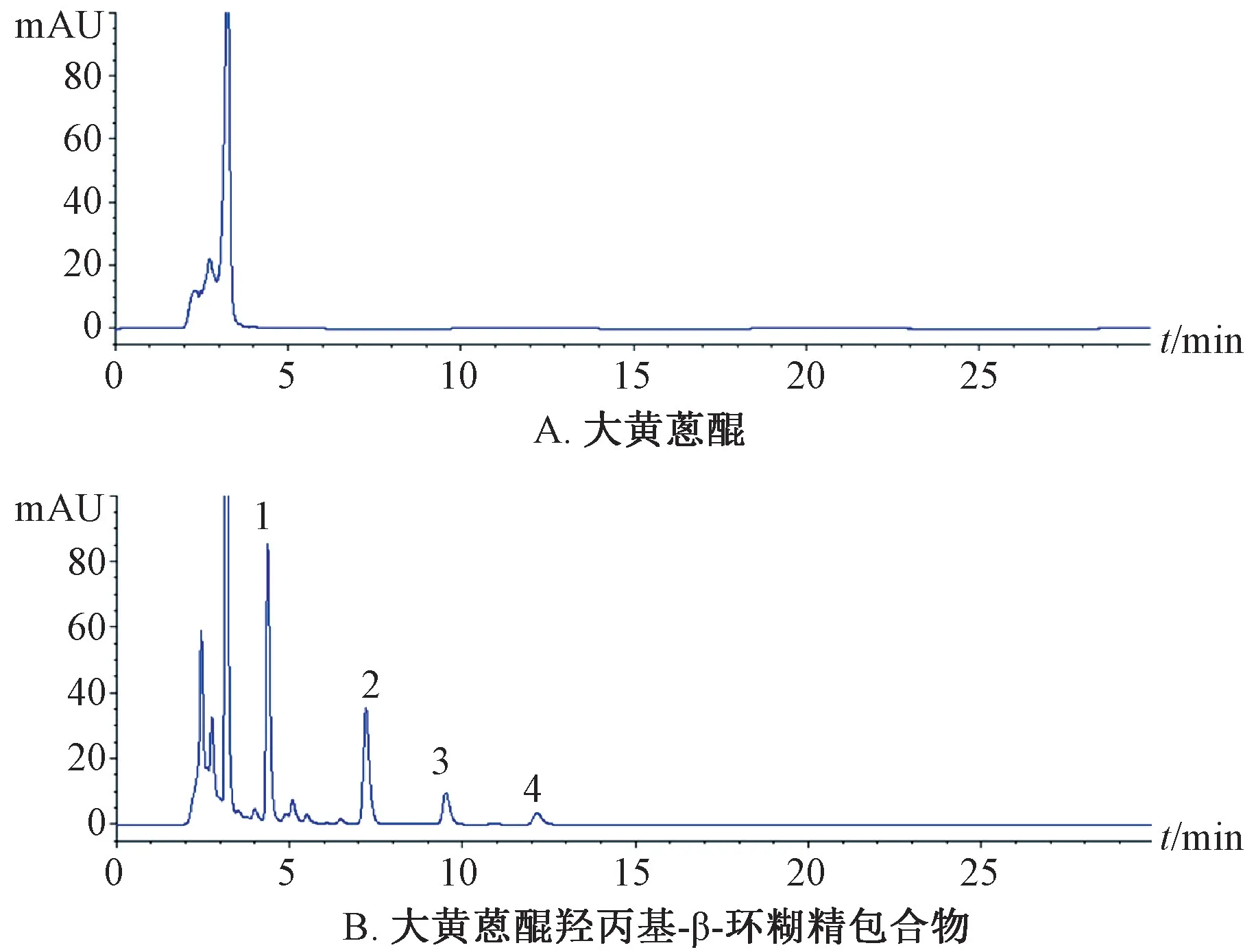
图4 各成分HPLC 色谱图
大黄作为泻下通腑的代表药,具有较好的通便导滞、活血祛瘀功效[20]。近年来,大黄蒽醌被不断开发,新功效也越来越多[21],但该成分在水中的溶解度低,渗透差,体内生物利用度不理想,从而限制了其临床应用。目前,有研究通过制备脂质体、聚合物胶束、纳米粒等方式来改善大黄蒽醌的水溶性,但存在工艺复杂、需使用有毒试剂等问题[22],而包合物的制备无需使用有毒试剂,并可大大改善药物溶解性和理化稳定性[23-24]。由于大黄蒽醌包合物在制备过程中需同时考虑各游离蒽醌的包合率、包合物得率等指标,故采用层次分析法来确定其权重系数。
在单因素试验过程中,本实验考察了大黄蒽醌与羟丙基-β-环糊精比例、包合时间、包合温度、搅拌速度,发现随着大黄蒽醌与羟丙基-β-环糊精比例不断增加,包合物综合评分也不断升高,但考虑到制剂成本及可行性,最终确定为1∶40。在正交试验过程中,本实验考察了包合时间、包合温度、搅拌速度对包合物制备工艺的影响,最终确定了最优工艺,其操作简便,稳定可行,适合工业化大生产,也可为后续原料药开发利用提供参考。然后,采用红外光谱、X 射线衍射、扫描电镜对包合物进行表征,发现大黄蒽醌已被包合入羟丙基-β-环糊精空穴中,并且其水溶性明显改善。
综上所述,本实验在单因素试验基础上采用正交试验优化大黄蒽醌羟丙基-β-环糊精包合工艺,可较好地增加该成分水溶性,为其后期应用提供参考,但其生物利用度能否提高尚不明确,仍需作进一步研究。
猜你喜欢包合物丙基蒽醌大孔吸附树脂纯化决明子总蒽醌工艺中成药(2018年10期)2018-10-26超声辅助双水相提取大黄中蒽醌类成分天然产物研究与开发(2018年8期)2018-09-10鸦胆子油β-环糊精包合物的制备中成药(2018年8期)2018-08-29石榴鞣花酸-羟丙基-β-环糊精包合物的制备中成药(2018年6期)2018-07-11鱼腥草挥发油HPCD包合物肠用温敏凝胶的制备中成药(2018年5期)2018-06-06大黄总蒽醌提取物对脑缺血再灌注损伤的保护作用及其机制中成药(2018年4期)2018-04-26N-丁氧基丙基-S-[2-(肟基)丙基]二硫代氨基甲酸酯浮选孔雀石的疏水机理中国有色金属学报(2018年2期)2018-03-26莪术油聚合环糊精包合物制备工艺的优化中成药(2017年12期)2018-01-19鱼腥草挥发油羟丙基-β环糊精包合物的制备中成药(2017年5期)2017-06-133-叠氮基丙基-β-D-吡喃半乳糖苷的合成工艺改进合成化学(2015年9期)2016-01-17本文来源:http://www.zhangdahai.com/shiyongfanwen/qitafanwen/2023/0920/657206.html